Automating your workflow: Nextflow and nf-core pipelines¶
Last updated: November 28, 2023
Section Overview
🕰 Time Estimation: X minutes
💬 Learning Objectives:
- Understand what is a pipeline/workflow and the Nextflow language.
- Learn about existing automated workflows from the bioinformatics community.
- Learn how to use the nf-core pipeline for bulk RNAseq analysis.
Workflows and pipelines¶
A pipeline consists of a chain of processing elements (processes, threads, coroutines, functions, etc.), arranged so that the output of each element is the input of the next; the name is by analogy to a physical pipeline. Narrowly speaking, a pipeline is linear and one-directional, though sometimes the term is applied to more general flows. For example, a primarily one-directional pipeline may have some communication in the other direction, known as a return channel or backchannel, or a pipeline may be fully bi-directional. Flows with one-directional tree and directed acyclic graph topologies behave similarly to (linear) pipelines – the lack of cycles makes them simple – and thus may be loosely referred to as "pipelines".
In our case, a "preprocessing" pipeline consists on concatenating all the processes explained in the previous lesson so that we have one continuous workflow from raw sequencing reads to a count matrix. For example, the RNASeq pipeline developed by the nf-core community (see below).
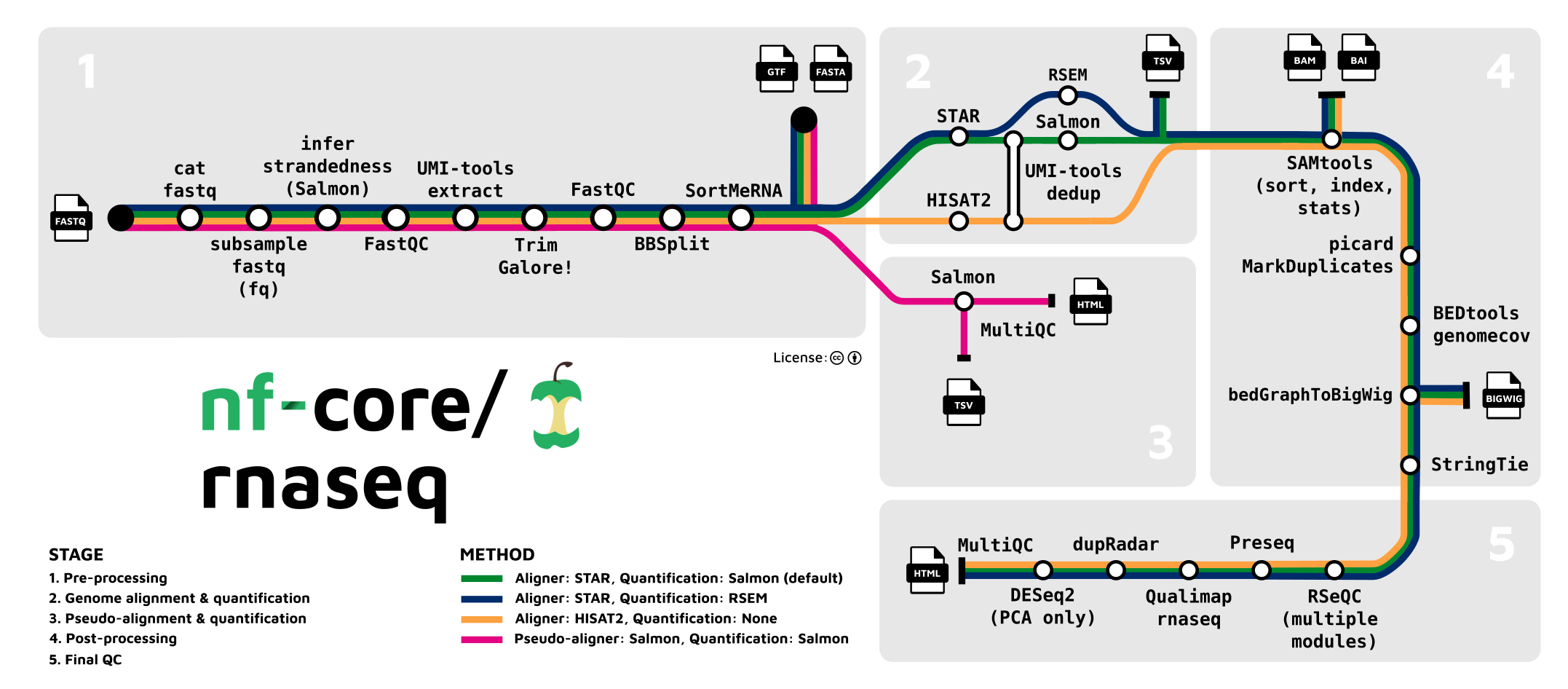
As you can see in the image above, each process, such as read trimming, QC, alignment, etc, are connected to each other. A workflow created in this way is ideal to reproduce analysis and makes the task of analysing RNAseq data much much easier.
Nextflow pipelines¶

Nextflow is a bioinformatics workflow manager that enables the development of portable and reproducible workflows. It supports deploying workflows on a variety of execution platforms including local, HPC schedulers, AWS Batch, Google Cloud Life Sciences, and Kubernetes. Additionally, it provides support for manage your workflow dependencies through built-in support for Conda, Spack, Docker, Podman, Singularity, Modules, and more.
With the rise of big data, techniques to analyse and run experiments on large datasets are increasingly necessary. Parallelization and distributed computing are the best ways to tackle this problem, but the tools commonly available to the bioinformatics community often lack good support for these techniques, or provide a model that fits badly with the specific requirements in the bioinformatics domain and, most of the time, require the knowledge of complex tools or low-level APIs.
Nextflow framework is based on the dataflow programming model, which greatly simplifies writing parallel and distributed pipelines without adding unnecessary complexity and letting you concentrate on the flow of data, i.e. the functional logic of the application/algorithm.
It doesn't aim to be another pipeline scripting language yet, but it is built around the idea that the Linux platform is the lingua franca of data science, since it provides many simple command line and scripting tools, which by themselves are powerful, but when chained together facilitate complex data manipulations.
In practice, this means that a Nextflow script is defined by composing many different processes. Each process can execute a given bioinformatics tool or scripting language, to which is added the ability to coordinate and synchronize the processes execution by simply specifying their inputs and outputs.
Features¶
- Fast prototyping: Nextflow allows you to write a computational pipeline by making it simpler to put together many different tasks. You may reuse your existing scripts and tools and you don't need to learn a new language or API to start using it.
- Reproducibility: Nextflow supports Docker and Singularity containers technology. This, along with the integration of the GitHub code sharing platform, allows you to write self-contained pipelines, manage versions and to rapidly reproduce any former configuration.
- Portable: Nextflow provides an abstraction layer between your pipeline's logic and the execution layer, so that it can be executed on multiple platforms without it changing. It provides out of the box executors for GridEngine, SLURM, LSF, PBS, Moab and HTCondor batch schedulers and for Kubernetes, Amazon AWS, Google Cloud and Microsoft Azure platforms.
- Unified parallelism: Nextflow is based on the dataflow programming model which greatly simplifies writing complex distributed pipelines. Parallelisation is implicitly defined by the processes input and output declarations. The resulting applications are inherently parallel and can scale-up or scale-out, transparently, without having to adapt to a specific platform architecture.
- Continuous checkpoints: All the intermediate results produced during the pipeline execution are automatically tracked. This allows you to resume its execution, from the last successfully executed step, no matter what the reason was for it stopping.
The nf-core project¶
![]()
The nf-core project is a community effort to collect a curated set of analysis pipelines built using Nextflow, an incredibly powerful and flexible workflow language. This means that all the tools and steps used in your RNAseq workflow can be automated and easily reproduced by other researchers if necessary. In addition, if you use any of the nf-core pipelines, you will be sure that all the necessary tools are available to you in any computer platform (Cloud computing, HPC or your personal computer).
The RNAseq pipeline enables using many different tools, such as STAR, RSEM, HISAT2 or Salmon, and allows quantification of gene/isoform counts and provides extensive quality control checks at each step of the workflow. We encourage your to take a look at the pipeline and its documentation if you need to preprocess your RNAseq reads from stratch, as well as checkout this introductory video.
nf-core/rnaseq: Usage for version 3.11.2¶
In this section, we will see some of the most important arguments that the pipeline uses to run a RNAseq preprocessing workflow. There are many more options for advanced users and we really encourage you to check them out at the official webpage. Below you will find the arguments we will use for our own data.
Running the pipeline¶
The typical command for running the pipeline is as follows:
nextflow run nf-core/rnaseq --input samplesheet.csv --outdir <OUTDIR> --genome GRCh37 -profile docker
This will launch the pipeline with the docker configuration profile. See below for more information about profiles.
Note that the pipeline will create the following files in your working directory:
work # Directory containing the nextflow working files
results # Finished results (configurable, see below)
.nextflow_log # Log file from Nextflow
# Other nextflow hidden files, eg. history of pipeline runs and old logs.
Info
- Options with a single hyphen are part of Nextflo, i.e.
-profile. - Pipeline specific parameters use a double-hyphen, i.e.
--input.
Core Nextflow arguments¶
-work-dir¶
Use this parameter to choose a path where the work folder, which containing all the intermediary files from the pipeline, should be saved.
-profile¶
Use this parameter to choose a configuration profile. Profiles can give configuration presets for different compute environments.
Several generic profiles are bundled with the pipeline which instruct the pipeline to use software packaged using different methods (Docker, Singularity, Podman, Shifter, Charliecloud, Conda) - see below. When using Biocontainers, most of these software packaging methods pull Docker containers from quay.io e.g FastQC except for Singularity which directly downloads Singularity images via https hosted by the Galaxy project and Conda which downloads and installs software locally from Bioconda.
Tip
It is highly recommended to use Docker or Singularity containers for full pipeline reproducibility, however when this is not possible, Conda is also supported.
The pipeline also dynamically loads configurations from https://github.com/nf-core/configs when it runs, making multiple config profiles for various institutional clusters available at run time. For more information and to see if your system is available in these configs please see the nf-core/configs documentation.
Note that multiple profiles can be loaded, for example: -profile test,docker - the order of arguments is important! They are loaded in sequence, so later profiles can overwrite earlier profiles.
If -profile is not specified, the pipeline will run locally and expect all software to be installed and available on the PATH. This is not recommended.
docker- A generic configuration profile to be used with Docker
singularity- A generic configuration profile to be used with Singularity
podman- A generic configuration profile to be used with Podman
shifter- A generic configuration profile to be used with Shifter
charliecloud- A generic configuration profile to be used with Charliecloud
conda- A generic configuration profile to be used with Conda. Please only use Conda as a last resort i.e. when it's not possible to run the pipeline with Docker, Singularity, Podman, Shifter or Charliecloud.
test- A profile with a complete configuration for automated testing
- Includes links to test data so needs no other parameters
-name¶
Use this parameter to give a unique name to the run. This name cannot be used again for another run in the same folder. This is very useful to track different runs since otherwise Nextflow will assign a random unique name to the run.
-resume¶
Specify this when restarting a pipeline. Nextflow will used cached results from any pipeline steps where the inputs are the same, continuing from where it got to previously.
You can also supply a run name to resume a specific run: -resume [run-name]. Use the nextflow log command to show previous run names.
-r¶
It is a good idea to specify a pipeline version when running the pipeline on your data. This ensures that a specific version of the pipeline code and software are used when you run your pipeline. If you keep using the same tag, you'll be running the same version of the pipeline, even if there have been changes to the code since.
First, go to the nf-core/rnaseq releases page and find the latest version number (numeric only), e.g., 1.3.1. Then specify this when running the pipeline with -r (one hyphen), e.g., -r 1.3.1.
This version number will be logged in reports when you run the pipeline, so that you'll know what you used when you look back in the future.
RNAseq pipeline arguments¶
Samplesheet input¶
You will need to create a samplesheet with information about the samples you would like to analyse before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 4 columns, and a header row as shown in the examples below.
Multiple runs of the same sample
The sample identifiers have to be the same when you have re-sequenced the same sample more than once e.g. to increase sequencing depth. The pipeline will concatenate the raw reads before performing any downstream analysis. Below is an example for the same sample sequenced across 3 lanes:
sample,fastq_1,fastq_2,strandedness
CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,unstranded
CONTROL_REP1,AEG588A1_S1_L003_R1_001.fastq.gz,AEG588A1_S1_L003_R2_001.fastq.gz,unstranded
CONTROL_REP1,AEG588A1_S1_L004_R1_001.fastq.gz,AEG588A1_S1_L004_R2_001.fastq.gz,unstranded
Full samplesheet
The pipeline will auto-detect whether a sample is single- or paired-end using the information provided in the samplesheet. The samplesheet can have as many columns as you desire, however, there is a strict requirement for the first 4 columns to match those defined in the table below.
A final samplesheet file consisting of both single- and paired-end data may look something like the one below. This is for 6 samples, where TREATMENT_REP3 has been sequenced twice.
sample,fastq_1,fastq_2,strandedness
CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,forward
CONTROL_REP2,AEG588A2_S2_L002_R1_001.fastq.gz,AEG588A2_S2_L002_R2_001.fastq.gz,forward
CONTROL_REP3,AEG588A3_S3_L002_R1_001.fastq.gz,AEG588A3_S3_L002_R2_001.fastq.gz,forward
TREATMENT_REP1,AEG588A4_S4_L003_R1_001.fastq.gz,,reverse
TREATMENT_REP2,AEG588A5_S5_L003_R1_001.fastq.gz,,reverse
TREATMENT_REP3,AEG588A6_S6_L003_R1_001.fastq.gz,,reverse
TREATMENT_REP3,AEG588A6_S6_L004_R1_001.fastq.gz,,reverse
| Column | Description |
|---|---|
sample | Custom sample name. This entry will be identical for multiple sequencing libraries/runs from the same sample. Spaces in sample names are automatically converted to underscores (_). |
fastq_1 | Full path to FastQ file for Illumina short reads 1. File has to be gzipped and have the extension ".fastq.gz" or ".fq.gz". |
fastq_2 | Full path to FastQ file for Illumina short reads 2. File has to be gzipped and have the extension ".fastq.gz" or ".fq.gz". |
strandedness | Sample strand-specificity. Must be one of unstranded, forward or reverse. |
Info
The group and replicate columns were replaced with a single sample column as of v3.1 of the pipeline. The sample column is essentially a concatenation of the group and replicate columns, however it now also offers more flexibility in instances where replicate information is not required e.g. when sequencing clinical samples. If all values of sample have the same number of underscores, fields defined by these underscore-separated names may be used in the PCA plots produced by the pipeline, to regain the ability to represent different groupings.
Results folder¶
The output directory where the results of the pipeline will be saved.
Alignment options¶
By default, the pipeline uses STAR (i.e. --aligner star_salmon) to map the raw FastQ reads to the reference genome, project the alignments onto the transcriptome and to perform the downstream BAM-level quantification with Salmon. STAR is fast but requires a lot of memory to run, typically around 38GB for the Human GRCh37 reference genome. Since the RSEM (i.e. --aligner star_rsem) workflow in the pipeline also uses STAR you should use the HISAT2 aligner (i.e. --aligner hisat2) if you have memory limitations.
You also have the option to pseudo-align and quantify your data with Salmon by providing the --pseudo_aligner salmon parameter. Salmon will then be run in addition to the standard alignment workflow defined by --aligner, mainly because it allows you to obtain QC metrics with respect to the genomic alignments. However, you can provide the --skip_alignment parameter if you would like to run Salmon in isolation.
Two additional parameters --extra_star_align_args and --extra_salmon_quant_args were added in v3.10 of the pipeline that allow you to append any custom parameters to the STAR align and Salmon quant commands, respectively. Note, the --seqBias and --gcBias are not provided to Salmon quant by default so you can provide these via --extra_salmon_quant_args '--seqBias --gcBias' if required.
Reference genome files¶
The minimum reference genome requirements are a FASTA and GTF file, all other files required to run the pipeline can be generated from these files. However, it is more storage and compute friendly if you are able to re-use reference genome files as efficiently as possible. It is recommended to use the --save_reference parameter if you are using the pipeline to build new indices (e.g. those unavailable on AWS iGenomes) so that you can save them somewhere locally. The index building step can be quite a time-consuming process and it permits their reuse for future runs of the pipeline to save disk space. You can then either provide the appropriate reference genome files on the command-line via the appropriate parameters (e.g. --star_index '/path/to/STAR/index/') or via a custom config file.
- If
--genomeis provided then the FASTA and GTF files (and existing indices) will be automatically obtained from AWS-iGenomes unless these have already been downloaded locally in the path specified by--igenomes_base. - If
--gffis provided as input then this will be converted to a GTF file, or the latter will be used if both are provided. - If
--gene_bedis not provided then it will be generated from the GTF file. - If
--additional_fastais provided then the features in this file (e.g. ERCC spike-ins) will be automatically concatenated onto both the reference FASTA file as well as the GTF annotation before building the appropriate indices.
When using --aligner star_rsem, both the STAR and RSEM indices should be present in the path specified by --rsem_index (see #568)
Note
Compressed reference files are also supported by the pipeline i.e. standard files with the .gz extension and indices folders with the tar.gz extension.
Process skipping options¶
There are several options to skip various steps within the workflow.
--skip_bigwig: Skip bigWig file creation--skip_stringtie: Skip StringTie.--skip_fastqc: Skip FastQC.--skip_preseq: Skip Preseq.--skip_dupradar: Skip dupRadar.--skip_qualimap: Skip Qualimap.--skip_rseqc: Skip RSeQC.--skip_biotype_qc: Skip additional featureCounts process for biotype QC.--skip_deseq2_qc: Skip DESeq2 PCA and heatmap plotting.--skip_multiqc: Skip MultiQC.--skip_qc: Skip all QC steps except for MultiQC.
Understanding the results folder¶
The pipeline will save everything inside the --outdir folder. Inside it you will find different results. In this section we will go through the most relevant results for this workshop. If you are interested in the full documentation, visit the nf-core rnaseq output docs.
1. pipeline_info¶
First, we will check the pipeline_info folder. Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.
pipeline_info/
- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter's are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv.
2. genome¶
A number of genome-specific files are generated by the pipeline because they are required for the downstream processing of the results. If the --save_reference parameter is provided then these will be saved in the genome/ directory. It is recommended to use the --save_reference parameter if you are using the pipeline to build new indices so that you can save them somewhere locally. The index building step can be quite a time-consuming process and it permits their reuse for future runs of the pipeline to save disk space.
Info
If you have not selected --save_reference, you will instead get a README.txt file containing information about the reference that was used for the run.
genome/
*.fa,*.gtf,*.gff,*.bed,.tsv: If the--save_referenceparameter is provided then all of the genome reference files will be placed in this directory
genome/index/
star/: Directory containing STAR indices.hisat2/: Directory containing HISAT2 indices.rsem/: Directory containing STAR and RSEM indices.salmon/: Directory containing Salmon indices.
3. multiqc¶
Results generated by MultiQC collate pipeline QC from supported tools i.e. FastQC, Cutadapt, SortMeRNA, STAR, RSEM, HISAT2, Salmon, SAMtools, Picard, RSeQC, Qualimap, Preseq and featureCounts. Additionally, various custom content has been added to the report to assess the output of dupRadar, DESeq2 and featureCounts biotypes, and to highlight samples failing a mimimum mapping threshold or those that failed to match the strand-specificity provided in the input samplesheet. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability.
multiqc/
multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing individual plots from the different tools used in the pipeline.
4. fastqc¶
The FastQC plots in this directory are generated relative to the raw, input reads. They may contain adapter sequence and regions of low quality.
fastqc/
*_fastqc.html: FastQC report containing quality metrics.*_fastqc.zip: Zip archive containing the FastQC report, tab-delimited data file and plot images.
5. trimgalore¶
In this folder you will find your trimmed and filtered fastq files from TrimGalore, including its fastqc results!
trimgalore/
*.fq.gz: If--save_trimmedis specified, FastQ files after adapter trimming will be placed in this directory.*_trimming_report.txt: Log file generated by Trim Galore!.
trimgalore/fastqc/
*_fastqc.html: FastQC report containing quality metrics for read 1 (and read2 if paired-end) after adapter trimming.*_fastqc.zip: Zip archive containing the FastQC report, tab-delimited data file and plot images.
6. aligner¶
Depending on the aligner that you use, this folder may change its contents and name. Generally, here you can find the aligned reads in .bam format, samtool stats, duplicated stats and different QC tools related to mapped reads.
Also, depending on the --aligner option you can either find the quantification results from either salmon (pseudoquantification) or rsem (traditional quantification, i.e., a count matrix)
From star_salmon
star_salmon/
*.Aligned.out.bam: If--save_align_intermedsis specified the original BAM file containing read alignments to the reference genome will be placed in this directory.
star_salmon/log/
*.SJ.out.tab: File containing filtered splice junctions detected after mapping the reads.*.Log.final.out: STAR alignment report containing the mapping results summary.*.Log.outand*.Log.progress.out: STAR log files containing detailed information about the run. Typically only useful for debugging purposes.
star_salmon/salmon/
salmon.merged.gene_counts.tsv: Matrix of gene-level raw counts across all samples.salmon.merged.gene_tpm.tsv: Matrix of gene-level TPM values across all samples.salmon.merged.gene_counts.rds: RDS object that can be loaded in R that contains a SummarizedExperiment container with the TPM (abundance), estimated counts (counts) and transcript length (length) in the assays slot for genes.salmon.merged.gene_counts_scaled.tsv: Matrix of gene-level scaled counts across all samples.salmon.merged.gene_counts_scaled.rds: RDS object that can be loaded in R that contains a SummarizedExperiment container with the TPM (abundance), estimated counts (counts) and transcript length (length) in the assays slot for genes.salmon.merged.gene_counts_length_scaled.tsv: Matrix of gene-level length-scaled counts across all samples.salmon.merged.gene_counts_length_scaled.rds: RDS object that can be loaded in R that contains a SummarizedExperiment container with the TPM (abundance), estimated counts (counts) and transcript length (length) in the assays slot for genes.salmon.merged.transcript_counts.tsv: Matrix of isoform-level raw counts across all samples.salmon.merged.transcript_tpm.tsv: Matrix of isoform-level TPM values across all samples.salmon.merged.transcript_counts.rds: RDS object that can be loaded in R that contains a SummarizedExperiment container with the TPM (abundance), estimated counts (counts) and transcript length (length) in the assays slot for transcripts.salmon_tx2gene.tsv: Tab-delimited file containing gene to transcripts ids mappings.
star_salmon/salmon/<SAMPLE>/
logs/: Contains the filesalmon_quant.loggiving a record of Salmon's quantification.quant.genes.sf: Salmon gene-level quantification of the sample, including feature length, effective length, TPM, and number of reads.quant.sf: Salmon transcript-level quantification of the sample, including feature length, effective length, TPM, and number of reads.
From star_rsem
star_rsem/
rsem.merged.gene_counts.tsv: Matrix of gene-level raw counts across all samples.rsem.merged.gene_tpm.tsv: Matrix of gene-level TPM values across all samples.rsem.merged.transcript_counts.tsv: Matrix of isoform-level raw counts across all samples.rsem.merged.transcript_tpm.tsv: Matrix of isoform-level TPM values across all samples.*.genes.results: RSEM gene-level quantification results for each sample.*.isoforms.results: RSEM isoform-level quantification results for each sample.
star_rsem/<SAMPLE>.stat/
*.cnt,*.model,*.theta: RSEM counts and statistics for each sample.
star_rsem/log/
*.log: STAR alignment report containing the mapping results summary.
7. pseudoaligner¶
This is the same output as the star_salmon/salmon folder.
Parts of this lesson have been taken from Wikipedia, the Nextflow webpage and the nf-core project webpage.
Created: November 28, 2023