Mapping and SNP Calling Tutorial¶
High-throughput sequencing technologies have in the past few years been producing millions of reads of human genome and other species. To be useful, this genetic information has to be 'put together' in a smart way, in the same way as the pieces of a puzzle (reads) need to be mounted according to a picture (reference genome).
The present tutorial, like the rest of the course material, is available at our open-source github repository, and is based on Kasper Munk's course repository.
Content¶
In this exercise section you will be exposed to different softwares used for mapping reads to a reference sequence and calling variants from the produced alignments. We will use a dataset composed of 28 individuals from 3 different regions: Africa, EastAsia and WestEurasia. You can find the metadata file, containing sample IDs and some extra information for each sample here: ../../Data/metadata/Sample_meta_subset.tsv
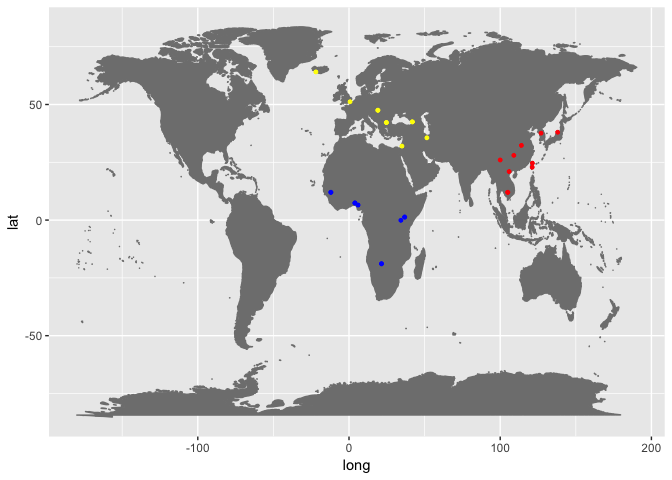
This dataset is a subset of the Simons Diversity Project. The following tutorial is based on the individual S_Ju_hoan_North-3, which is an african sample from Namibia.
How to make this notebook work¶
- In this notebook we will use primarily
Randcommand line bashcommands. - This notebook integrates multiple coding languages. This means you need to choose a kernel every time we shift from one language to another. A kernel contains a programming language and the necessary packages to run the course material. To choose a kernel, go on the menu on the top of the page and select
Kernel --> Change Kernel, and then select the preferred one. We will shift between two kernels, and along the code in this notebook you will see a picture telling you to change kernel. The two pictures are below:
 Shift to the
Shift to the Bash kernel
 Shift to the
Shift to the popgen course kernel
- You can run the code in each cell by clicking on the run cell sign in the toolbar, or simply by pressing Shift+Enter. When the code is done running, a small green check sign will appear on the left side.
- You need to run the cells in sequential order to execute the analysis. Please do not run a cell until the one above is done running, and do not skip any cells
- The code goes along with textual descriptions to help you understand what happens. Please try not to focus on understanding the code itself in too much detail - but rather try to focus on the explanations and on the output of the commands
- You can create new code cells by pressing
+in the Menu bar above or by pressing B after you select a cell.
Learning outcomes¶
At the end of this tutorial you will be able to
- handle and understand the various steps needed to prepare data for alignment using standard tools like
samtools - apply genome alignment tools (
burrows-wheeler aligner BWAandMinimap2) and comment their outcome - make sense of the various data formats (
fasta,fastq,sam/bam,vcf, ...) - explore and analyze data on a genome viewer
Mapping reads against the reference¶
The first step when dealing with raw reads is mapping (aligning) them to a reference sequence. We will use two different aligners for mapping the reads against the reference genome; one is the classical Burrows-Wheeler aligner (BWA), and the other is the newer and faster Minimap2. Minimap2 is especially faster (up to 50X faster) and as accurate as BWA in aligning long reads, but works also well with short read data like ours.
Two input files are needed to do genome mapping:
- Fasta file containing your reference genome Which was downloaded from hg19. We need to copy the file from our
Datafolder, since it is a protected folder where we cannot write any file. - The reads in fastq format, which can be found in
Data/fastq/. Here we will not need to copy the files to another folder, because we will only read them.
We create a link to Data, so it is easier to access it. Then we make a folder called Reference and copy the file chr2.fa in it. Finally we create a folder where to save our results.
Choose the Bash kernel
mkdir -p ../../Reference
cp ../../Data/fasta/chr2.fa ../../Reference/
ln -s ../../Data
ln -s ../../Reference
mkdir -p Results
ln: failed to create symbolic link './Data': File exists
Alignment with BWA¶
BWA allows for fast and accurate alignment of short reads to an indexed reference sequence. Here is the manual. We will focus on a 10 MB region of chromosome 2 (from 135MB to 145MB), which contains the lactase gene.
First we need to index the reference file for later use. This creates index files used to perform the alignment. To produce these files, run the following command. Here, -a bwtsw specifies the indexing algorithm; specifically bwtsw which is capable of handling large genomes such as the human genome (the standard option is -a is, which cannot handle beyond 2MB genomes).
bwa index -a bwtsw Reference/chr2.fa
[bwa_index] Pack FASTA... 1.49 sec [bwa_index] Construct BWT for the packed sequence... [BWTIncCreate] textLength=486398746, availableWord=46224508 [BWTIncConstructFromPacked] 10 iterations done. 75165770 characters processed. [BWTIncConstructFromPacked] 20 iterations done. 139904490 characters processed. [BWTIncConstructFromPacked] 30 iterations done. 197440426 characters processed. [BWTIncConstructFromPacked] 40 iterations done. 248574506 characters processed. [BWTIncConstructFromPacked] 50 iterations done. 294018586 characters processed. [BWTIncConstructFromPacked] 60 iterations done. 334405370 characters processed. [BWTIncConstructFromPacked] 70 iterations done. 370297226 characters processed. [BWTIncConstructFromPacked] 80 iterations done. 402193962 characters processed. [BWTIncConstructFromPacked] 90 iterations done. 430539834 characters processed. [BWTIncConstructFromPacked] 100 iterations done. 455729658 characters processed. [BWTIncConstructFromPacked] 110 iterations done. 478114410 characters processed. [bwt_gen] Finished constructing BWT in 115 iterations. [bwa_index] 123.49 seconds elapse. [bwa_index] Update BWT... 1.26 sec [bwa_index] Pack forward-only FASTA... 0.94 sec [bwa_index] Construct SA from BWT and Occ... 57.30 sec [main] Version: 0.7.17-r1188 [main] CMD: bwa index -a bwtsw Reference/chr2.fa [main] Real time: 199.439 sec; CPU: 184.484 sec
The resulting bwa index files are in the same folder Reference as the reference genome file.
ls Reference
chr2.fa chr2.fa.amb chr2.fa.ann chr2.fa.bwt chr2.fa.pac chr2.fa.sa
We also need to generate a fasta file index, which contains the genomic coordinates of the reference file. This can be done using the samtools program faidx. Again, the resulting index files are in the Reference folder:
samtools faidx Reference/chr2.fa
ls Reference
chr2.fa chr2.fa.ann chr2.fa.fai chr2.fa.sa chr2.fa.amb chr2.fa.bwt chr2.fa.pac
Now you can map the reads to the reference. This will take a few minutes. Note how the alignment command bwa mem is piped with the symbol | at the end of the command. This allows the output of the alignment with bwa mem not to be written anywhere, but to be transferred (piped) directly into the following command samtools sort, which sorts the aligned genome by coordinates and creates a file in bam format. The sorting is necessary as the output of bwa mem is unsorted, and sorted files are usually the standard for genomic analysis. The piping speeds up the running time of the commands, avoiding to write intermediate files, and thus saving also disk space.
bwa mem -t 16 -p Reference/chr2.fa './fastq/S_Ju_hoan_North-3.region.fq' | samtools sort -O BAM -o './Results/S_Ju_hoan_North-3.bam'
[M::bwa_idx_load_from_disk] read 0 ALT contigs [M::process] read 1600642 sequences (160000012 bp)... [M::process] 1586946 single-end sequences; 13696 paired-end sequences [M::process] read 1600786 sequences (160000003 bp)... [M::mem_process_seqs] Processed 1586946 reads in 230.885 CPU sec, 29.368 real sec [M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (4, 710, 0, 0) [M::mem_pestat] skip orientation FF as there are not enough pairs [M::mem_pestat] analyzing insert size distribution for orientation FR... [M::mem_pestat] (25, 50, 75) percentile: (74, 86, 95) [M::mem_pestat] low and high boundaries for computing mean and std.dev: (32, 137) [M::mem_pestat] mean and std.dev: (82.89, 15.62) [M::mem_pestat] low and high boundaries for proper pairs: (11, 158) [M::mem_pestat] skip orientation RF as there are not enough pairs [M::mem_pestat] skip orientation RR as there are not enough pairs [M::mem_process_seqs] Processed 13696 reads in 3.202 CPU sec, 0.380 real sec [M::process] 1588148 single-end sequences; 12638 paired-end sequences [M::process] read 642285 sequences (64201734 bp)... [M::mem_process_seqs] Processed 1588148 reads in 203.147 CPU sec, 26.514 real sec [M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (1, 757, 0, 1) [M::mem_pestat] skip orientation FF as there are not enough pairs [M::mem_pestat] analyzing insert size distribution for orientation FR... [M::mem_pestat] (25, 50, 75) percentile: (73, 85, 95) [M::mem_pestat] low and high boundaries for computing mean and std.dev: (29, 139) [M::mem_pestat] mean and std.dev: (81.95, 16.02) [M::mem_pestat] low and high boundaries for proper pairs: (7, 161) [M::mem_pestat] skip orientation RF as there are not enough pairs [M::mem_pestat] skip orientation RR as there are not enough pairs [M::mem_process_seqs] Processed 12638 reads in 2.745 CPU sec, 0.351 real sec [M::process] 637205 single-end sequences; 5080 paired-end sequences [M::mem_process_seqs] Processed 637205 reads in 91.616 CPU sec, 12.521 real sec [M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (0, 300, 0, 0) [M::mem_pestat] skip orientation FF as there are not enough pairs [M::mem_pestat] analyzing insert size distribution for orientation FR... [M::mem_pestat] (25, 50, 75) percentile: (74, 86, 94) [M::mem_pestat] low and high boundaries for computing mean and std.dev: (34, 134) [M::mem_pestat] mean and std.dev: (82.85, 14.79) [M::mem_pestat] low and high boundaries for proper pairs: (14, 154) [M::mem_pestat] skip orientation RF as there are not enough pairs [M::mem_pestat] skip orientation RR as there are not enough pairs [M::mem_process_seqs] Processed 5080 reads in 1.123 CPU sec, 0.116 real sec [main] Version: 0.7.17-r1188 [main] CMD: bwa mem -t 16 -p Reference/chr2.fa ./fastq/S_Ju_hoan_North-3.region.fq [main] Real time: 94.034 sec; CPU: 535.906 sec [bam_sort_core] merging from 1 files and 1 in-memory blocks...
Bam files follow this structure, where each element is tab-separated inside the output file:

- Read name: ID for the given read.
- Flags: Combination of bitwise FLAGs that provide information on how the read is mapped Extra information.
- Position: Chromosome and position of the first base in the alignment.
- MAPQ: Probability of wrong mapping of the read. It's in Phred scale, so higher numbers mean lower probabilities:
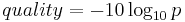
- CIGAR: summary of the alignment, including start position on the reference sequence, matches, mismatches, deletions and insertions. It may also include information on soft/hard clipping, i.e bases in the 3' and 5' ends of teh read that are not part of the alignment. Extra information
- Mate information: chromosome and start position of teh read pair, and inferred insert size.
- Quality scores: base qualities of the read.
- Metadata: optional extra information.
For more information, read this
Have a look at the bam file generated and compare it with the structure above. There is an error message, but do not mind it.
samtools view ./Results/S_Ju_hoan_North-3.bam | head -n5
HS2000-690_130:3:2115:19816:65272 16 2 48688 0 100M * 0 0 TCATGTTCTTTGCAGAGACACGAATGGAGCTGGAATCCATTATCTTCAGCAAACTAACACAGGAACAGAAAACCAAACACTGCATGTTCTCACTCATAAG CCDDCCEAEEFFFFFFHGHHGJJJIJJIJJJJJJGJJIGJJJJJIJJIJJIIGIJJIGHJJJJJJJJIJJIHJIIIHGDJIJIHFIIHHHHFFFFFFCCC NM:i:8 MD:Z:6A8G4T1G12G44C0A12T5 AS:i:60 XS:i:59 HS2000-690_130:4:2202:2325:9318 16 2 126500 0 100M * 0 0 CTCAGTGCTCAATGGTGCCCAGGCTGGAGTGCAGTGGCGTGATCTCGGCTCGCTACAACCTCCACCTCCCAGCCGCCTGCCTTGGCCTCCCAAAGTGCCG DCCA83DDCDDC?B<<8DBAABCC>9DCC@CA@BB?<BCAA?55??;/@?@;;>BC?5B;(A=-HGDGGF@7BHGGDGGHGGEC@GJHHHHHFFFDD@@@ NM:i:0 MD:Z:100 AS:i:100 XS:i:100 HS2000-690_130:2:1311:9371:14324 0 2 127285 0 100M * 0 0 GAAAAATTCTTCTGCCTTGGGATCCTGTTGATCTATGACCTTACCCCCAACCCTGTGCTCTCTGAAACATGTGCTGTGTCCACTCAGGGTTAAATGGATT @@CFFFFFGHHHHJJJGJIFHIIJJJIIJGEGIJGFHFIGIIJIIIIJJJIJJJJJGCGIIJJJ;@EHG>E?EEC?@CBBDFEEEDAABCA2>(,5>:AC NM:i:0 MD:Z:100 AS:i:100 XS:i:100 HS2000-690_130:3:2307:1163:23064 0 2 130911 0 41M59S * 0 0 TCTTCTTGCTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGGGGGGAGTTGTTAAAGTTTTTTCTCATCCCTTTTAAAATTTTTTTAAAATTTTTA CCCFFFFFHHHDHJJIIGIJJIJJJHFDDDDDDDBDDDDB############################################################ NM:i:1 MD:Z:2C38 AS:i:38 XS:i:38 HS2000-690_130:4:2206:11079:24232 0 2 299169 4 100M * 0 0 ATCCTAAGCAAAAAGAACAAAGCTGGAGGCATCATGTTACCTGACTTCAAACTATACTACAAGGCTACAGTAACCAAAACAGCATGGTACTGGTACCAAA CCCFFFDFGGHGGJJJJEGGIJIIIIEIJFBFGGGHEGGIGBHGGIGIJJIHIIJJJJJIIJJJJFEHGIHEEEH=?DFFDEA@ECBADDCCA>ACDDCA NM:i:2 MD:Z:82T16T0 AS:i:94 XS:i:90 samtools view: writing to standard output failed: Broken pipe samtools view: error closing standard output: -1
You can get useful statistics using samtools flagstat
samtools flagstat ./Results/S_Ju_hoan_North-3.bam
3845680 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 secondary 1967 + 0 supplementary 0 + 0 duplicates 3809175 + 0 mapped (99.05% : N/A) 31414 + 0 paired in sequencing 15707 + 0 read1 15707 + 0 read2 3724 + 0 properly paired (11.85% : N/A) 4014 + 0 with itself and mate mapped 13699 + 0 singletons (43.61% : N/A) 0 + 0 with mate mapped to a different chr 0 + 0 with mate mapped to a different chr (mapQ>=5)
Alignment with Minimap2¶
Minimap2 is a versatile sequence alignment program that aligns DNA or mRNA sequences against a large reference database. Typical use cases include: (1) mapping PacBio or Oxford Nanopore genomic reads to the human genome; (2) finding overlaps between long reads with error rate up to ~15%; (3) splice-aware alignment of PacBio Iso-Seq or Nanopore cDNA or Direct RNA reads against a reference genome; (4) aligning Illumina single- or paired-end reads; (5) assembly-to-assembly alignment; (6) full-genome alignment between two closely related species with divergence below ~15%. For >100bp Illumina short reads, minimap2 is three times as fast as BWA-MEM and Bowtie2, and as accurate on simulated data.
We apply minimap2 to the same data used for bwa. Here, the option -x sr is used to select a preset for a specific type of data. In our case, sr denotes Short single-end reads without splicing. More information about other alignment scenarios can be found in the manual.
minimap2 -a -x sr Reference/chr2.fa fastq/S_Ju_hoan_North-3.region.fq | \
samtools sort -O BAM -o './Results/S_Ju_hoan_North-3.minimap.bam'
[M::mm_idx_gen::6.499*1.00] collected minimizers [M::mm_idx_gen::7.354*1.23] sorted minimizers [M::main::7.354*1.23] loaded/built the index for 1 target sequence(s) [M::mm_mapopt_update::7.354*1.23] mid_occ = 1000 [M::mm_idx_stat] kmer size: 21; skip: 11; is_hpc: 0; #seq: 1 [M::mm_idx_stat::7.762*1.22] distinct minimizers: 34516711 (96.91% are singletons); average occurrences: 1.154; average spacing: 6.108; total length: 243199373 [M::worker_pipeline::18.784*2.30] mapped 500234 sequences [M::worker_pipeline::25.132*2.52] mapped 500191 sequences [M::worker_pipeline::32.092*2.66] mapped 500194 sequences [M::worker_pipeline::37.964*2.74] mapped 500202 sequences [M::worker_pipeline::43.456*2.80] mapped 500257 sequences [M::worker_pipeline::66.683*2.45] mapped 500275 sequences [M::worker_pipeline::67.986*2.41] mapped 500203 sequences [M::worker_pipeline::68.832*2.39] mapped 342157 sequences [M::main] Version: 2.24-r1122 [M::main] CMD: minimap2 -a -x sr Reference/chr2.fa fastq/S_Ju_hoan_North-3.region.fq [M::main] Real time: 68.928 sec; CPU: 164.602 sec; Peak RSS: 2.686 GB [bam_sort_core] merging from 1 files and 1 in-memory blocks...
Look at the output on the screen from bwa mem and minimap2. Both the real time (the actual time you wait for the command to run) and the CPU time (how much time in total multiple CPUs have been working while you waited for the real time) should be lower for minimap2, since this is in general faster than bwa mem.
Explore the results with the IGV software¶
To visualize the alignment of the reads to the reference genome we have just produced, we will use the Integrative Genome Visualizer IGV. You can run it from your laptop after downloading it, or running it through uCloud from the Genomics Sandbox application (only if you are an uCloud user). But first, we need to generate an index file for this software to work. Indexing a genome sorted BAM file allows one to quickly extract alignments overlapping particular genomic regions. Moreover, indexing is required by genome viewers such as IGV so that the viewers can quickly display alignments in each genomic region to which you navigate.
samtools index ./Results/S_Ju_hoan_North-3.bam
IGV is an Integrative Genomics viewer and can be very useful to look at the results of Mapping and SNP calling. Three files are necessary to look at this dataset: the reference sequence and the
.bam and .bai files in the Results folder. Since we are using a human reference, the sequence is already available in the software and does not need a download. Download the .bam and .bai files on your laptop, and if you want to use uCloud, upload them into one of your personal folders.
Reading in the data¶
To select the reference genome, go to Genomes ---> Load from server... ---> Filter by human and choose the Human hg19 reference.
Now, you want to see the chromosomes and genes: download the mapping results by going to File ---> Load from File... ----> S_Ju_hoan_North-3.bam.
When you zoom in to the lactase region on chromosome 2 (chr2:136,545,410-136,594,750), you will see something like this: 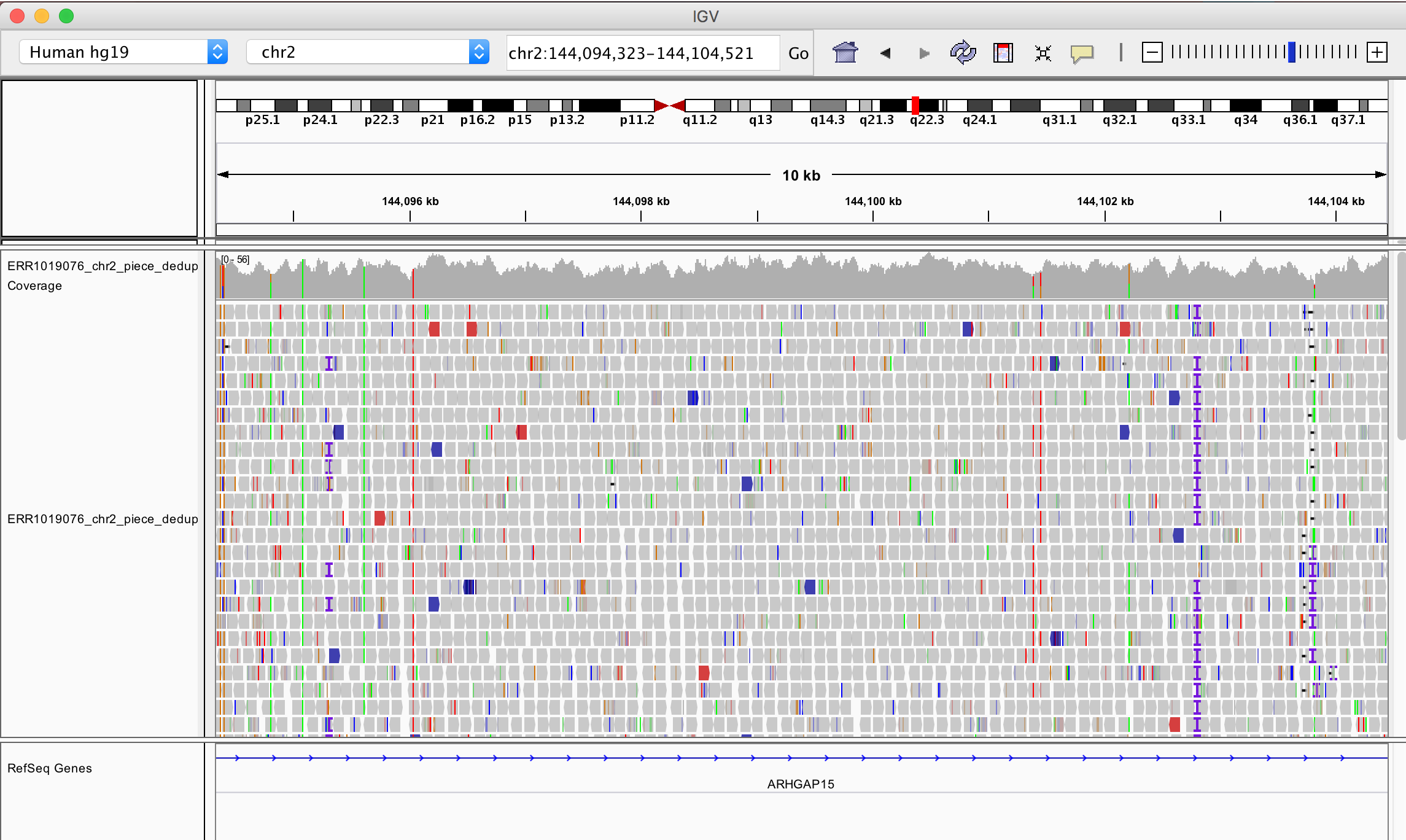
Try to understand what are the different attributes present in the viewer. If you zoom in very much you will find single nucleotide polymorphisms (SNPs), where the reference sequence does not have the same nucleotide as the data mapped to.
Analyzing read coverage¶
One of the attributes one could learn from mapping reads back to the reference is the coverage of reads across the genome. In order to calculate the coverage depth you can use the command samtools depth.
samtools depth Results/S_Ju_hoan_North-3.bam > Results/S_Ju_hoan_North-3.coverage
less Results/S_Ju_hoan_North-3.coverage | head
2 48688 1 2 48689 1 2 48690 1 2 48691 1 2 48692 1 2 48693 1 2 48694 1 2 48695 1 2 48696 1 2 48697 1
We now switch to the popgen course kernel and use the R language to look at the depth file
library(ggplot2)
library(dplyr)
Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
scaf <- read.table("./Results/S_Ju_hoan_North-3.coverage", header=FALSE, sep="\t",
na.strings="NA", dec=".", strip.white=TRUE,
col.names = c("Scaffold", "locus", "depth"))
head(scaf)
| Scaffold | locus | depth | |
|---|---|---|---|
| <int> | <int> | <int> | |
| 1 | 2 | 48688 | 1 |
| 2 | 2 | 48689 | 1 |
| 3 | 2 | 48690 | 1 |
| 4 | 2 | 48691 | 1 |
| 5 | 2 | 48692 | 1 |
| 6 | 2 | 48693 | 1 |
We plot the average coverage in windows of 100 loci and save the plot in Results/S_Ju_hoan_North-3.coverage.pdf
# Average in windows
scaf %>%
mutate(rounded_position = round(locus, -2)) %>%
group_by(rounded_position) %>%
summarize(mean_cov = mean(depth)) -> compressed
# Plotting the data
p <- ggplot(data = compressed, aes(x=rounded_position, y=mean_cov)) + geom_area() + theme_classic() + ylim(0, 400)
# Saving your coverage plot
ggsave("Results/S_Ju_hoan_North-3.coverage.pdf",p)
Saving 6.67 x 6.67 in image
Look at the pdf with the plot of the coverage. What are the conclusions you can extract from these analysis? Does the coverage match with what you observed with IGV? Does it match with what you would expect, i.e what you know from the data?
SNP calling¶
Even though just a tiny portion (around 2%) of our genomes are based of protein coding regions, this partition contains most of the disease causal variants (mutations), and that is why variant calling is so important from a medical point of view. From the population genetics side, it is also possible to use these variants to establish differences between individuals, populations and species. It can also be used to clarify the genetic basis of adaptation.
Once we have mapped our reads we can now start with variant detection. We will use the software bcftools, a program for variant calling and manipulating files in the Variant Call Format (VCF) and its binary counterpart BCF.
You will be using the VCF format further in the course, for now let's just count the number of heterozygous SNPs in each individual:
Choose the Bash kernel
bcftools mpileup --threads 16 -Ou --skip-indels -f Reference/chr2.fa Results/S_Ju_hoan_North-3.bam | \
bcftools call -mv -Ov -o Results/S_Ju_hoan_North-3.vcf
Note: none of --samples-file, --ploidy or --ploidy-file given, assuming all sites are diploid [mpileup] 1 samples in 1 input files
The output is a single VCF file containing all the variants that bcftools identified.
Look at the output vcf file. What does the format look like? Does that match with what you observed in the IGV? Download the VCF file to the IGV browser to look atthe SNPs.
Here we can count how many line the file contains
less ./Results/S_Ju_hoan_North-3.vcf | wc -l
15793
Note that part of those lines (in this case 28) are just descriptions of the meaning of each column.
less ./Results/S_Ju_hoan_North-3.vcf | head -n 30
##fileformat=VCFv4.2 ##FILTER=<ID=PASS,Description="All filters passed"> ##bcftoolsVersion=1.8+htslib-1.8 ##bcftoolsCommand=mpileup --threads 16 -Ou --skip-indels -f Reference/chr2.fa Results/S_Ju_hoan_North-3.bam ##reference=file://Reference/chr2.fa ##contig=<ID=2,length=243199373> ##ALT=<ID=*,Description="Represents allele(s) other than observed."> ##INFO=<ID=INDEL,Number=0,Type=Flag,Description="Indicates that the variant is an INDEL."> ##INFO=<ID=IDV,Number=1,Type=Integer,Description="Maximum number of reads supporting an indel"> ##INFO=<ID=IMF,Number=1,Type=Float,Description="Maximum fraction of reads supporting an indel"> ##INFO=<ID=DP,Number=1,Type=Integer,Description="Raw read depth"> ##INFO=<ID=VDB,Number=1,Type=Float,Description="Variant Distance Bias for filtering splice-site artefacts in RNA-seq data (bigger is better)",Version="3"> ##INFO=<ID=RPB,Number=1,Type=Float,Description="Mann-Whitney U test of Read Position Bias (bigger is better)"> ##INFO=<ID=MQB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality Bias (bigger is better)"> ##INFO=<ID=BQB,Number=1,Type=Float,Description="Mann-Whitney U test of Base Quality Bias (bigger is better)"> ##INFO=<ID=MQSB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality vs Strand Bias (bigger is better)"> ##INFO=<ID=SGB,Number=1,Type=Float,Description="Segregation based metric."> ##INFO=<ID=MQ0F,Number=1,Type=Float,Description="Fraction of MQ0 reads (smaller is better)"> ##FORMAT=<ID=PL,Number=G,Type=Integer,Description="List of Phred-scaled genotype likelihoods"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##INFO=<ID=ICB,Number=1,Type=Float,Description="Inbreeding Coefficient Binomial test (bigger is better)"> ##INFO=<ID=HOB,Number=1,Type=Float,Description="Bias in the number of HOMs number (smaller is better)"> ##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes for each ALT allele, in the same order as listed"> ##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes"> ##INFO=<ID=DP4,Number=4,Type=Integer,Description="Number of high-quality ref-forward , ref-reverse, alt-forward and alt-reverse bases"> ##INFO=<ID=MQ,Number=1,Type=Integer,Description="Average mapping quality"> ##bcftools_callVersion=1.8+htslib-1.8 ##bcftools_callCommand=call -mv -Ov -o Results/S_Ju_hoan_North-3.vcf; Date=Tue Jan 3 15:05:21 2023 #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Results/S_Ju_hoan_North-3.bam 2 5282534 . T A 5.04598 . DP=1;SGB=-0.379885;MQ0F=0;AC=2;AN=2;DP4=0,0,1,0;MQ=60 GT:PL 1/1:33,3,0
The called genotype is written in the last column (together with other values) and can be
- 0/0 - the sample is homozygous to the reference (note that these sites usually won't be listed in single sample vcf files as they are not variants)
- 0/1 OR 0/1 - the sample is heterozygous, carrying 1 copy of each of the REF and ALT alleles
- 1/1 - the sample is homozygous for the alternate allele
We can count how many of those different called genotype there are by using grep. For the homozigous alternate allele,
grep '1/1' ./Results/S_Ju_hoan_North-3.vcf | wc -l
5444
For the homozigous genotypes
grep '0/1\|1/0' ../../Results/S_Ju_hoan_North-3.vcf | wc -l
10308
Choose the popgen course kernel
We can read in the vcf file in R
t = read.table("../../Results/S_Ju_hoan_North-3.vcf",header=FALSE, sep="\t", na.strings="NA", dec=".", strip.white=TRUE)
head(t)
| V1 | V2 | V3 | V4 | V5 | V6 | V7 | V8 | V9 | V10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| <int> | <int> | <chr> | <chr> | <chr> | <dbl> | <chr> | <chr> | <chr> | <chr> | |
| 1 | 2 | 5282534 | . | T | A | 5.04598 | . | DP=1;SGB=-0.379885;MQ0F=0;AC=2;AN=2;DP4=0,0,1,0;MQ=60 | GT:PL | 1/1:33,3,0 |
| 2 | 2 | 8015657 | . | C | G | 4.38466 | . | DP=1;SGB=-0.379885;MQ0F=0;AC=2;AN=2;DP4=0,0,0,1;MQ=54 | GT:PL | 1/1:32,3,0 |
| 3 | 2 | 10349722 | . | C | T | 17.49230 | . | DP=2;VDB=0.06;SGB=-0.453602;MQSB=1;MQ0F=0;AC=2;AN=2;DP4=0,0,1,1;MQ=24 | GT:PL | 1/1:47,6,0 |
| 4 | 2 | 12297549 | . | T | C | 4.38466 | . | DP=1;SGB=-0.379885;MQ0F=0;AC=2;AN=2;DP4=0,0,0,1;MQ=40 | GT:PL | 1/1:32,3,0 |
| 5 | 2 | 13959931 | . | T | G | 18.47640 | . | DP=3;VDB=0.0312982;SGB=-0.511536;MQSB=1;MQ0F=0;AC=2;AN=2;DP4=0,0,2,1;MQ=19 | GT:PL | 1/1:48,9,0 |
| 6 | 2 | 13959944 | . | T | C | 24.42990 | . | DP=3;VDB=0.0433485;SGB=-0.511536;MQSB=1;MQ0F=0;AC=2;AN=2;DP4=0,0,2,1;MQ=19 | GT:PL | 1/1:54,9,0 |
Choose column number 10
geno_column = t[['V10']]
geno_column[1:10]
- '1/1:33,3,0'
- '1/1:32,3,0'
- '1/1:47,6,0'
- '1/1:32,3,0'
- '1/1:48,9,0'
- '1/1:54,9,0'
- '1/1:48,3,0'
- '1/1:53,9,0'
- '1/1:30,1,0'
- '1/1:33,3,0'
For each element in the vector, choose only the part of text before :, so that the genotype is isolated from other values
geno = sapply(geno_column, function(x) unlist(strsplit(x,':'))[1])
geno[1:10]
- 1/1:33,3,0
- '1/1'
- 1/1:32,3,0
- '1/1'
- 1/1:47,6,0
- '1/1'
- 1/1:32,3,0
- '1/1'
- 1/1:48,9,0
- '1/1'
- 1/1:54,9,0
- '1/1'
- 1/1:48,3,0
- '1/1'
- 1/1:53,9,0
- '1/1'
- 1/1:30,1,0
- '1/1'
- 1/1:33,3,0
- '1/1'
Given this information you are now able to estimate the mean heterozygosity for your individual of the 10 MB region in chromosome 2.
1 - (sum(geno=='0/1')/length(geno))^2 - (sum(geno=='1/1')/length(geno))^2