GWAS Quality Control using PLINK¶
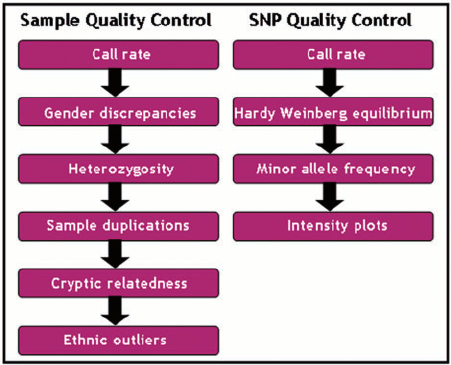
One of the most important aspects of a Genome Wide Analysis Studies (GWAS) is to do thorough Quality control (QC) of the data before analysing it. The QC of GWAS goes through various steps (see figure below), and we will illustrate some of them in this tutorial.
How to make this notebook work¶
- In this notebook we will use both the
command line bashcommands andRto setup the file folders. - Having to shift between two languages, you need to choose a kernel every time we shift from one language to another. A kernel contains a programming language and the necessary packages to run the course material. To choose a kernel, go on the menu on the top of the page and select
Kernel --> Change Kernel, and then select the preferred one. We will shift between two kernels, and along the code in this notebook you will see a picture telling you to change kernel. The two pictures are below:
 Shift to the
Shift to the Bash kernel
 Shift to the
Shift to the popgen course kernel
- You can run the code in each cell by clicking on the run cell sign in the toolbar, or simply by pressing Shift+Enter. When the code is done running, a small green check sign will appear on the left side.
- You need to run the cells in sequential order to execute the analysis. Please do not run a cell until the one above is done running, and do not skip any cells
- The code goes along with textual descriptions to help you understand what happens. Please try not to focus on understanding the code itself in too much detail - but rather try to focus on the explanations and on the output of the commands
- You can create new code cells by pressing
+in the Menu bar above or by pressing B after you select a cell.
Background material¶
Anderson et al 2010, review on data quality control for GWAS studies (on which this tutorial is based).
A video lecture on quality control for GWAS.
Uffelmann et al 2021, a review on GWAS methods.
Software:¶
We will be using plink, a comprehensive tool for handling and analyzing SNP data that can perform many different kinds of analyses. Check the documentation here
If you want info about a specific command you can also use help command:
plink --help <name_of_command>Data:¶
In this practical, we will go through the steps in performing quality control (QC) of genotype data from a simulated genome-wide association study of 1000 cases and 1000 controls, typed for 317,503 autosomal and X chromosome SNPs.
We will link to the following folder, which contains the necessary files in .bed, .bim, .fam formats, and make the Results folder:
Shift to the Bash kernel
ln -s ../../Data/GWAS/GWAS_QC_data/
mkdir -p Results
ln: failed to create symbolic link './GWAS_QC_data': File exists
Sample QC¶
We will begin by performing sample QC, including calculation of call rates, heterozygosity, and sex discordance. We will then perform SNP QC, including calculation of call rates and deviation from Hardy-Weinberg equilibrium.
The following command will infer the sex of the sample by looking at the mean homozygosity rate across X-chromosome markers and compares it to the sex stated in the .fam file.
plink --bfile GWAS_QC_data/GWA-data --check-sex --out Results/GWA-QC
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC.log. Options in effect: --bfile GWAS_QC_data/GWA-data --check-sex --out Results/GWA-QC 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 2000 people (997 males, 1003 females) loaded from .fam. 2000 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 2000 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11440 het. haploid genotypes present (see Results/GWA-QC.hh ); many commands treat these as missing. Total genotyping rate is 0.985682. 317503 variants and 2000 people pass filters and QC. Among remaining phenotypes, 1023 are cases and 977 are controls. --check-sex: 8921 Xchr and 0 Ychr variant(s) scanned, 3 problems detected. Report written to Results/GWA-QC.sexcheck .
Q1) Take a look at the output file GWA-data.sexcheck. How many problematic samples are there?
Problematic samples can be removed by copying the family ID (FID) and individual ID (IID) of the samples into a text file and using the remove command. Below, we use head to see how the file GWA-data.sexcheck is structured. The column STATUS might contain the indicator we need to filter out problematic samples, so we apply grep -v to see which lines do not contain the string OK. Then we write those lines in the file wrong_sex.txt so we can apply filtering with plink afterwards.
#preview of the file
head Results/GWA-QC.sexcheck
FID IID PEDSEX SNPSEX STATUS F 1 1 1 1 OK 0.9552 2 2 1 1 OK 0.9997 3 3 1 1 OK 0.9935 4 4 1 1 OK 1 5 5 2 2 OK 0.007004 6 6 2 2 OK 0.007528 7 7 2 2 OK -0.003711 8 8 2 2 OK 0.007389 9 9 2 2 OK -0.009868
#check lines without "OK"
grep -v "OK" Results/GWA-QC.sexcheck
FID IID PEDSEX SNPSEX STATUS F 772 772 2 0 PROBLEM 0.3084 853 853 2 0 PROBLEM 0.3666 1920 1920 2 0 PROBLEM 0.4066
#put those lines in a file
grep -v "OK" Results/GWA-QC.sexcheck > Results/wrong_sex.txt
cat Results/wrong_sex.txt
FID IID PEDSEX SNPSEX STATUS F 772 772 2 0 PROBLEM 0.3084 853 853 2 0 PROBLEM 0.3666 1920 1920 2 0 PROBLEM 0.4066
Alternatively, one can ose the awk command to print the elements in the fifth column of the file (awk '{if (NR > 1) print $5}'), and then sort those elements alphabetically (sort -k1,1) and print their unique occurrences (option -u), so that it can be decided which use of grep -v makes sense. We can see there are only two possible values of STATUS: OK, PROBLEM.
awk '{if (NR > 1) print $5}' Results/GWA-QC.sexcheck | sort -k1,1 -u
OK PROBLEM
Now we run plink with the command --remove to filter out the problematic samples. The --out option in plink specifies the prefix of the output files that plink generates. And when we use the --make-bed command we are writing the output to the specified prefix. In this case, all our output files will have the prefix: GWA-QC.
plink --bfile GWAS_QC_data/GWA-data --remove Results/wrong_sex.txt --make-bed --out Results/GWA-QC_sflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt.log. Options in effect: --bfile GWAS_QC_data/GWA-data --make-bed --out Results/GWA-QC_sflt --remove Results/wrong_sex.txt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 2000 people (997 males, 1003 females) loaded from .fam. 2000 phenotype values loaded from .fam. --remove: 1997 people remaining. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1997 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11440 het. haploid genotypes present (see Results/GWA-QC_sflt.hh ); many commands treat these as missing. Total genotyping rate in remaining samples is 0.985924. 317503 variants and 1997 people pass filters and QC. Among remaining phenotypes, 1022 are cases and 975 are controls. --make-bed to Results/GWA-QC_sflt.bed + Results/GWA-QC_sflt.bim + Results/GWA-QC_sflt.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
Q2) Each time a plink command is run it writes a summary to a log file (the file name ends with .log). Look at the log file after removing the problematic individuals. How many cases and controls are left in the data set?
Identification of individuals with elevated missing data rates or outlying heterozygosity rate¶
The following plink command will create the files GWA-QC.imiss and GWA-QC.lmiss. The fourth column in the file GWA-data.imiss (N_MISS) denotes the number of missing SNPs and the sixth column (F_MISS) denotes the proportion of missing SNPs per individual.
plink --bfile Results/GWA-QC_sflt --missing --out Results/GWA-QC_sflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt.log. Options in effect: --bfile Results/GWA-QC_sflt --missing --out Results/GWA-QC_sflt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1997 people (997 males, 1000 females) loaded from .fam. 1997 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1997 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11440 het. haploid genotypes present (see Results/GWA-QC_sflt.hh ); many commands treat these as missing. Total genotyping rate is 0.985924. --missing: Sample missing data report written to Results/GWA-QC_sflt.imiss, and variant-based missing data report written to Results/GWA-QC_sflt.lmiss.
Now with plink we create the file GWA-data.het, in which the third column denotes the observed number of homozygous genotypes O(Hom) and the fifth column denotes the number of non-missing genotypes N(NM) per individual.
You can calculate the observed heterozygosity rate per individual using the formula:
$Het = \frac{N(NM) − O(Hom)}{N(NM)}$
plink --bfile Results/GWA-QC_sflt --het --out Results/GWA-QC_sflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt.log. Options in effect: --bfile Results/GWA-QC_sflt --het --out Results/GWA-QC_sflt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1997 people (997 males, 1000 females) loaded from .fam. 1997 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1997 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11440 het. haploid genotypes present (see Results/GWA-QC_sflt.hh ); many commands treat these as missing. Total genotyping rate is 0.985924. 317503 variants and 1997 people pass filters and QC. Among remaining phenotypes, 1022 are cases and 975 are controls. --het: 305281 variants scanned, report written to Results/GWA-QC_sflt.het .
Shift to the popgen course kernel
By running this I get the mean of number of missing genotypes and missing frequencies for each sample. I also compute the observed heterozygosity on each individual. Then, I plot these results, outlying extreme values on both variables, i.e missing frequency greater than 3% and heterozygosity lower or greaater than 3 standard deviations from the mean. A high proportion of missing genotypes is indicative of low DNA quality or concentration, while higher or lower proportion of heterozygotes is indicative of contamination or inbreeding, respectively.
Imiss = read.table('Results/GWA-QC_sflt.imiss', header = TRUE)
mean(Imiss$N_MISS)
mean(Imiss$F_MISS)
het = read.table('Results/GWA-QC_sflt.het', header = T)
het$het_cal = (het$N.NM. - het$O.HOM.)/ het$N.NM.
mean_het = mean(het$het_cal)
sd_het = sd(het$het_cal)
plot(x = het$het_cal,y = Imiss$F_MISS, lwd =4, col = 'light blue', xlab = 'Heterozygosity rate per individual', ylab = 'Proportion of missing SNPs per individual')
abline(h = 0.03, col = 'red', lty = 3)
abline(v = mean_het + 3*sd_het, col = 'red', lty = 3)
abline(v = mean_het - 3*sd_het, col = 'red', lty = 3)

Proceed to filter samples based on these criteria:
right_tail = mean_het + 3*sd(het$het_cal)
left_tail = mean_het - 3*sd(het$het_cal)
filtering = cbind(Imiss, het)
outlier_ind = subset(filtering, filtering$F_MISS >= 0.03 | filtering$het_cal > right_tail | filtering$het_cal < left_tail)
nrow(outlier_ind)
write.table(outlier_ind[,c(1,2)], 'Results/wrong_het_missing_values.txt', col.names = FALSE, row.names = FALSE)
Shift to the Bash kernel
plink --bfile Results/GWA-QC_sflt --remove Results/wrong_het_missing_values.txt --make-bed --out Results/GWA-QC_sflt_hflt_mflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt.log. Options in effect: --bfile Results/GWA-QC_sflt --make-bed --out Results/GWA-QC_sflt_hflt_mflt --remove Results/wrong_het_missing_values.txt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1997 people (997 males, 1000 females) loaded from .fam. 1997 phenotype values loaded from .fam. --remove: 1954 people remaining. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1954 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11116 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt.hh ); many commands treat these as missing. Total genotyping rate in remaining samples is 0.98671. 317503 variants and 1954 people pass filters and QC. Among remaining phenotypes, 995 are cases and 959 are controls. --make-bed to Results/GWA-QC_sflt_hflt_mflt.bed + Results/GWA-QC_sflt_hflt_mflt.bim + Results/GWA-QC_sflt_hflt_mflt.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
After that, you should have the same number of variants and 1954 samples left. You should also see that we have created bed, bim and fam files for GWA-QC_sflt_mflt_hflt, containing our filtered dataset.
ls Results/GWA-QC_sflt_hflt_mflt*
Results/GWA-QC_sflt_hflt_mflt.bed Results/GWA-QC_sflt_hflt_mflt.hh Results/GWA-QC_sflt_hflt_mflt.bim Results/GWA-QC_sflt_hflt_mflt.log Results/GWA-QC_sflt_hflt_mflt.fam
Identification of duplicated or related individuals¶
To identify duplicated or related individuals we will calculate the identity by descent (IBD) matrix, indicative of relatedness between individuals or repeated samples, that could bias our GWAS. This works best if it is done on a set of non-correlated SNPs. So first we will “prune” the data and create a list of SNPs where no pair (within a given genomic interval) has an r2 value greater than a given threshold, typically chosen to be 0.2. This can be done by the indep-pairwise command, using 500kb as window size and 5 variants as step size:
plink --bfile Results/GWA-QC_sflt_hflt_mflt --indep-pairwise 500kb 5 0.2 --out Results/GWA-QC_sflt_hflt_mflt
plink --bfile Results/GWA-QC_sflt_hflt_mflt --extract Results/GWA-QC_sflt_hflt_mflt.prune.in --genome --min 0.185 --out Results/GWA-QC_sflt_hflt_mflt
ls -lht Results/
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt --indep-pairwise 500kb 5 0.2 --out Results/GWA-QC_sflt_hflt_mflt Warning: --indep-pairwise step size should be 1 when window size is in kb units. 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1954 people (973 males, 981 females) loaded from .fam. 1954 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1954 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11116 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt.hh ); many commands treat these as missing. Total genotyping rate is 0.98671. 317503 variants and 1954 people pass filters and QC. Among remaining phenotypes, 995 are cases and 959 are controls. Pruned 210 variants from chromosome 1, leaving 23065. Pruned 222 variants from chromosome 2, leaving 25129. Pruned 234 variants from chromosome 3, leaving 21346. Pruned 180 variants from chromosome 4, leaving 18933. Pruned 184 variants from chromosome 5, leaving 19088. Pruned 277 variants from chromosome 6, leaving 20534. Pruned 169 variants from chromosome 7, leaving 16506. Pruned 208 variants from chromosome 8, leaving 18066. Pruned 134 variants from chromosome 9, leaving 15701. Pruned 145 variants from chromosome 10, leaving 15447. Pruned 134 variants from chromosome 11, leaving 14526. Pruned 159 variants from chromosome 12, leaving 14873. Pruned 117 variants from chromosome 13, leaving 11409. Pruned 61 variants from chromosome 14, leaving 9768. Pruned 64 variants from chromosome 15, leaving 8836. Pruned 71 variants from chromosome 16, leaving 8935. Pruned 60 variants from chromosome 17, leaving 8283. Pruned 100 variants from chromosome 18, leaving 10395. Pruned 90 variants from chromosome 19, leaving 5837. Pruned 77 variants from chromosome 20, leaving 7759. Pruned 55 variants from chromosome 21, leaving 5438. Pruned 104 variants from chromosome 22, leaving 5401. Pruned 249 variants from chromosome 23, leaving 8922. Pruned 0 variants from chromosome 25, leaving 2. Pruning complete. 3304 of 317503 variants removed. Marker lists written to Results/GWA-QC_sflt_hflt_mflt.prune.in and Results/GWA-QC_sflt_hflt_mflt.prune.out . PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt --extract Results/GWA-QC_sflt_hflt_mflt.prune.in --genome --min 0.185 --out Results/GWA-QC_sflt_hflt_mflt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1954 people (973 males, 981 females) loaded from .fam. 1954 phenotype values loaded from .fam. --extract: 314199 variants remaining. Using up to 63 threads (change this with --threads). Before main variant filters, 1954 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 11116 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt.hh ); many commands treat these as missing. Total genotyping rate is 0.996987. 314199 variants and 1954 people pass filters and QC. Among remaining phenotypes, 995 are cases and 959 are controls. Excluding 8922 variants on non-autosomes from IBD calculation. IBD calculations complete. Finished writing Results/GWA-QC_sflt_hflt_mflt.genome . total 334M -rw-r--r--. 1 ucloud users 1.9K Jan 18 09:56 GWA-QC_sflt_hflt_mflt.genome -rw-r--r--. 1 ucloud users 1.3K Jan 18 09:56 GWA-QC_sflt_hflt_mflt.log -rw-r--r--. 1 ucloud users 205K Jan 18 09:56 GWA-QC_sflt_hflt_mflt.hh -rw-r--r--. 1 ucloud users 33K Jan 18 09:56 GWA-QC_sflt_hflt_mflt.prune.out -rw-r--r--. 1 ucloud users 3.1M Jan 18 09:56 GWA-QC_sflt_hflt_mflt.prune.in -rw-r--r--. 1 ucloud users 573 Jan 18 09:51 GWA-data_sflt_hflt_mflt.log -rw-r--r--. 1 ucloud users 8.4M Jan 18 09:51 GWA-QC_sflt_hflt_mflt.bim -rw-r--r--. 1 ucloud users 33K Jan 18 09:51 GWA-QC_sflt_hflt_mflt.fam -rw-r--r--. 1 ucloud users 149M Jan 18 09:51 GWA-QC_sflt_hflt_mflt.bed -rw-r--r--. 1 ucloud users 372 Jan 18 09:49 wrong_het_missing_values.txt -rw-r--r--. 1 ucloud users 1.1K Jan 18 09:46 GWA-QC_sflt.log -rw-r--r--. 1 ucloud users 121K Jan 18 09:46 GWA-QC_sflt.het -rw-r--r--. 1 ucloud users 211K Jan 18 09:46 GWA-QC_sflt.hh -rw-r--r--. 1 ucloud users 94K Jan 18 09:43 GWA-QC_sflt.imiss -rw-r--r--. 1 ucloud users 14M Jan 18 09:43 GWA-QC_sflt.lmiss -rw-r--r--. 1 ucloud users 8.4M Jan 18 09:42 GWA-QC_sflt.bim -rw-r--r--. 1 ucloud users 33K Jan 18 09:42 GWA-QC_sflt.fam -rw-r--r--. 1 ucloud users 152M Jan 18 09:42 GWA-QC_sflt.bed -rw-r--r--. 1 ucloud users 248 Jan 18 09:40 wrong_sex.txt -rw-r--r--. 1 ucloud users 1.1K Jan 18 09:36 GWA-QC.log -rw-r--r--. 1 ucloud users 122K Jan 18 09:36 GWA-QC.sexcheck -rw-r--r--. 1 ucloud users 211K Jan 18 09:36 GWA-QC.hh
The --min 0.185 option means that it will only print the calculated IBD if it is above 0.185 (Mean between second-degree relatives:0.25 and third-degree relatives:0.125). The PI_HAT value in column 10 of the output file will be a number between 0 and 1 saying how much of the genome the two individuals share (1 for identical twins, 0.5 for siblings etc.). This command will produce a file called GWA-data_sflt_hflt_mflt.genome .
Shift to the popgen course kernel
In R, I proceed in analyzing the output file, named Results/GWA-QC_sflt_hflt_mflt.genome, to remove a member from each of the pairs that are too closely related from the data set.
ibd = read.table('Results/GWA-QC_sflt_hflt_mflt.genome', header = TRUE)
members = ibd[,1]
members = unique(members)
length(members)
write.table(cbind(members,members), file = 'Results/wrong_ibd.txt', col.names = F, row.names = F)
With these lines of code, I obtain a unique set of 14 samples that show outlying IDB with another sample. Here we are exploiting the fact that FID and IID are the same in our dataset, but we would have to contition on both if that wasn't the case. Back to the command line, I filter out samples in our dataset using this file by running:
Shift to the Bash kernel
plink --bfile Results/GWA-QC_sflt_hflt_mflt --remove Results/wrong_ibd.txt --make-bed --out Results/GWA-QC_sflt_hflt_mflt_iflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt_iflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt --make-bed --out Results/GWA-QC_sflt_hflt_mflt_iflt --remove Results/wrong_ibd.txt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1954 people (973 males, 981 females) loaded from .fam. 1954 phenotype values loaded from .fam. --remove: 1940 people remaining. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1940 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 10977 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt_iflt.hh ); many commands treat these as missing. Total genotyping rate in remaining samples is 0.986697. 317503 variants and 1940 people pass filters and QC. Among remaining phenotypes, 988 are cases and 952 are controls. --make-bed to Results/GWA-QC_sflt_hflt_mflt_iflt.bed + Results/GWA-QC_sflt_hflt_mflt_iflt.bim + Results/GWA-QC_sflt_hflt_mflt_iflt.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
I get a new set of files, containing the same number of variables and the samples that survived the wrong sex, the missing data and ibd filtering. At this point, you should have 1940 samples, 988 cases and 952 controls.
plink --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --missing --out Results/GWA-QC_sflt_hflt_mflt_iflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt_iflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --missing --out Results/GWA-QC_sflt_hflt_mflt_iflt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1940 people (965 males, 975 females) loaded from .fam. 1940 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1940 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 10977 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt_iflt.hh ); many commands treat these as missing. Total genotyping rate is 0.986697. --missing: Sample missing data report written to Results/GWA-QC_sflt_hflt_mflt_iflt.imiss, and variant-based missing data report written to Results/GWA-QC_sflt_hflt_mflt_iflt.lmiss.
I open Results/GWA-data_sflt_hflt_mflt_iflt.lmiss in R to plot an histogram of the variant's missing frequency:
Shift to the popgen course kernel
miss_data = read.table('Results/GWA-QC_sflt_hflt_mflt_iflt.lmiss', header = T)
hist(miss_data$F_MISS, breaks = 50, col = 'light blue', main = 'Missing Data Distribution', xlab = 'Missing Data rate')

A lot of care must be taken when filtering variants, since we can lose potential variants associaated with the phenotypic trait. Therefore, we only filter these variants in which the missigness is associated with the phenotype, i.e case or control, so they could be a source of bias in our study.
We use plink with the --test-missing command to test for association between missingness and case/control status, using Fisher's exact test. It produces a file with .missing suffix. We make a list of all the names of all SNPs where the differential missingness p-value is less than 1e-5. The list is saved as fail-diffmiss-qc.txt
Shift to the Bash kernel
plink --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --test-missing --out Results/GWA-QC_sflt_hflt_mflt_iflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt_iflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --out Results/GWA-QC_sflt_hflt_mflt_iflt --test-missing 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1940 people (965 males, 975 females) loaded from .fam. 1940 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1940 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 10977 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt_iflt.hh ); many commands treat these as missing. Total genotyping rate is 0.986697. 317503 variants and 1940 people pass filters and QC. Among remaining phenotypes, 988 are cases and 952 are controls. Writing --test-missing report to Results/GWA-QC_sflt_hflt_mflt_iflt.missing ... done.
This outputs a file named GWA-data_sflt_mflt_hflt_iflt.missing. Using R and applying a significance criterion of 10e-5, I get significant variants by:
Shift to the popgen course kernel
test_missing = read.table('Results/GWA-QC_sflt_hflt_mflt_iflt.missing', header = TRUE)
fail_diffmiss_qc = test_missing[test_missing$P < 10e-5, 2]
write.table(fail_diffmiss_qc, file = 'Results/fail-diffmiss-qc.txt', row.names = F, col.names = F)
Now we can remove low-quality SNPs. In addition to removing SNPs identified with differential call rates between cases and controls, this command removes SNPs with call rate less than 95% with --geno option and deviation from HWE (p<1e-5) with the --hwe option. It also removes all SNPs with minor allele frequency less than a specified threshold using the --maf option.
Shift to the Bash kernel
plink --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --exclude Results/fail-diffmiss-qc.txt --geno 0.05 --hwe 0.00001 --maf 0.01 --make-bed --out Results/GWA-QC_sflt_hflt_mflt_iflt_vflt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/ (C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to Results/GWA-QC_sflt_hflt_mflt_iflt_vflt.log. Options in effect: --bfile Results/GWA-QC_sflt_hflt_mflt_iflt --exclude Results/fail-diffmiss-qc.txt --geno 0.05 --hwe 0.00001 --maf 0.01 --make-bed --out Results/GWA-QC_sflt_hflt_mflt_iflt_vflt 385583 MB RAM detected; reserving 192791 MB for main workspace. 317503 variants loaded from .bim file. 1940 people (965 males, 975 females) loaded from .fam. 1940 phenotype values loaded from .fam. --exclude: 317503 variants remaining. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 1940 founders and 0 nonfounders present. Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done. Warning: 10977 het. haploid genotypes present (see Results/GWA-QC_sflt_hflt_mflt_iflt_vflt.hh ); many commands treat these as missing. Total genotyping rate is 0.986697. 3340 variants removed due to missing genotype data (--geno). Warning: --hwe observation counts vary by more than 10%, due to the X chromosome. You may want to use a less stringent --hwe p-value threshold for X chromosome variants. --hwe: 16 variants removed due to Hardy-Weinberg exact test. 251 variants removed due to minor allele threshold(s) (--maf/--max-maf/--mac/--max-mac). 313896 variants and 1940 people pass filters and QC. Among remaining phenotypes, 988 are cases and 952 are controls. --make-bed to Results/GWA-QC_sflt_hflt_mflt_iflt_vflt.bed + Results/GWA-QC_sflt_hflt_mflt_iflt_vflt.bim + Results/GWA-QC_sflt_hflt_mflt_iflt_vflt.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
Deviations from HWE might indicate genotype error, but we choose a very low siginifcance value, since deviations from HWE might also be a product of selection implying that we could be filtering out variants potentially associated with the phenotypic trait. Samples with low allele frequency are also more likely to be miss-calls. Additionally, these variants have low power to be significant associations in the GWAS study due to their low small sample size.
After all filtering, you should be left with:
- 313896 variants.
- 1940 people (988 cases and 952 controls).