Building genealogies¶
As seen in the selective sweep tutorial, male X chromosomes are easy to use for haplotype-based analysis, as they are haploid and therefore does not require phasing. However, autosomes are diploid, and we would like to use haplotype-based analysis here as well. This can be done through phasing. Multiple phasing methods are known, such as Beagle (already used in the previous tutorial about phasing) and Shapeit. If you are lucky, the dataset you are using has already been phased, which is where we start in this tutorial. We are back to limit us to looking at chr2. The data is taken from 60 individuals in the 1000Genomes project. In this dataset, there are 20 individuals from the following populations: GBR (Brits/Scots), JPT (Japanese), YRI (Yoruban).
Background material¶
We will use the software Relate to infer the genealogies from genome data. It is optimal to read the software article, and for those who want a deeper knowledge of it, the second software paper illustrating updates to the program is here.
How to make this notebook work¶
- In this notebook we will use only
command line Bash. You need to choose a kernel, that contains a programming language and the necessary packages to run the course material. To choose the right kernel, go on the menu on the top of the page and selectKernel --> Change Kernel, and then chooseBash. - You can run the code in each cell by clicking on the run cell sign in the toolbar, or simply by pressing Shift+Enter. When the code is done running, a small green check sign will appear on the left side.
- You need to run the cells in sequential order to execute the analysis. Please do not run a cell until the one above is done running, and do not skip any cells
- The code goes along with textual descriptions to help you understand what happens. Please try not to focus on understanding the code itself in too much detail - but rather try to focus on the explanations and on the output of the commands
- You can create new code cells by pressing
+in the Menu bar above or by pressing B after you select a cell.
Learning outcomes¶
At the end of this tutorial you will be able to
- Understand the background theory behind
relateand connect the theory to the application of this tutorial - Interpret and discuss the various steps for preparing and analyzing data in
relate - Compare and criticize the usage of multiple vs single populations in the inference of ancestral trees
- Explore, interpret and generalize simple simulated ancestral trees
Genealogies based on Genome-wide data¶
Knowledge of genome-wide genealogies for many individuals obviously allows for more robust evolutionary analyses for humans and other species - but the computational burden has been unfeasible for long. Relate is a fast and scalable (beyond 10,000 sequences) software that estimates branch lengths, mutational ages and variable historical population sizes, as well as allowing for data errors. Below is a simple scheme of how the software works - it essentially builds a matrix of distances, where closest sections of chromosomes are more probable to originate from a copying event. From this matrix, a coalescent tree is inferred with dedicated techniques.
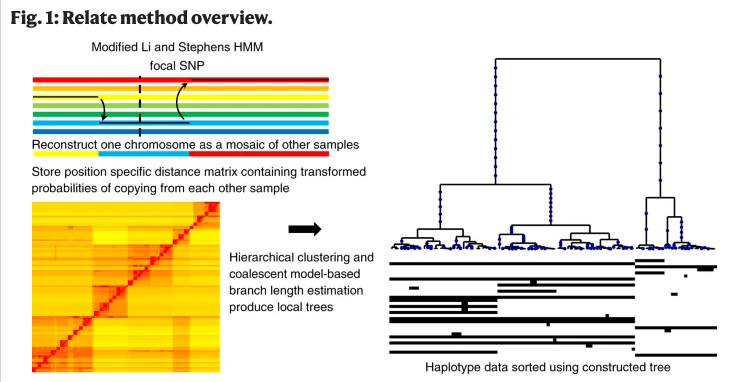
We link to the folder relate_data that containes various files needed for the analysis and to the folder containing the necessary software files. We create also the Results folder.
ln -s ../../Data/relate_data
ln -s ../../Data/software/relate_v1.1.9_x86_64_static relate
mkdir -p Results
ln: failed to create symbolic link './relate_data': File exists
For the analysis we use the following files:
20140520.chr2.strict_mask.fastais a mask containing areas that either have abnormal read depth or has been identified to contain repetitive elements.genetic_map_chr2_combined_b37.txtis a recombination map.human_ancestor_2.fais the ancestral state of every site, based on an alignment with Gorilla, Chimp and Human genomes.60_inds.txtis a metadata file detailing the population and region for each sample.chr2_130_145_phased.vcf.gzis the phased genotype vcf.
Relate does not accept the standard vcf file format, but instead uses a haps/sample format. You can read up on in the Relate documentation. The authors have been so kind as to supply a script to transform it.
Note that input files in Relate always have to be supplied without file extensions.
ls relate_data
20140520.chr2.strict_mask.fasta 50_GBR_inds.txt 50_GBR_sample_list.txt 60_inds.txt 60_sample_list.txt ALL.chr2.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz ALL.chr2.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz.tbi ALL.chr2.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz.vcfidx chr2_130_145_phased.vcf.gz chr2_GBR_phased.vcf.gz genetic_map_chr2_combined_b37.txt human_ancestor_2.fa
relate/bin/RelateFileFormats --mode ConvertFromVcf --haps Results/chr2.haps --sample Results/chr2.sample -i relate_data/chr2_130_145_phased
--------------------------------------------------------- Converting vcf file format to haps/sample file format.. Output written to Results/chr2.haps and Results/chr2.sample. CPU Time spent: 1.036033s; Max Memory usage: 2.508Mb. ---------------------------------------------------------
relate/scripts/PrepareInputFiles/PrepareInputFiles.sh --haps Results/chr2.haps --sample Results/chr2.sample \
--ancestor relate_data/human_ancestor_2.fa \
--mask relate_data/20140520.chr2.strict_mask.fasta \
-o Results/prep.chr2
******************************** Parameters passed to script: haps = Results/chr2.haps sample = Results/chr2.sample ancestor = relate_data/human_ancestor_2.fa output = Results/prep.chr2 mask = relate_data/20140520.chr2.strict_mask.fasta ******************************** --------------------------------------------------------- Removing non-biallelic SNPs.. Removed 0 non-biallelic SNPs. Output written to Results/prep.chr2_biall.haps. CPU Time spent: 0.060300s; Max Memory usage: 0.92Mb. --------------------------------------------------------- --------------------------------------------------------- Detemining ancestral allele and flipping SNPs if necessary... Had to remove 10485 SNPs because of non-matching nucleotides Number of flipped SNPs is 12662. Output written to Results/prep.chr2_ancest.haps. CPU Time spent: 1.447338s; Max Memory usage: 410.672Mb. --------------------------------------------------------- relate/scripts/PrepareInputFiles/PrepareInputFiles.sh: line 120: file: command not found --------------------------------------------------------- Filtering haps using mask.. Removed 19650 SNPs. Output written to Results/prep.chr2_filtered.haps and Results/prep.chr2_filtered.dist CPU Time spent: 1.484666s; Max Memory usage: 412.308Mb. ---------------------------------------------------------
Look at the outputs above, and answer to the following questions.
- Q1: How many SNPs were removed due to non-matching nucleotides?
- Q2: How many were removed due to the mask?
Estimation of ancestral trees¶
Now, the input is fully prepared, and the inference of genealogies with Relate can run. This should take less than a minute.
relate/bin/Relate --mode All -m 1.25e-8 -N 30000 --haps Results/prep.chr2.haps.gz --sample Results/prep.chr2.sample.gz \
--map relate_data/genetic_map_chr2_combined_b37.txt \
-o chr2_relate
********************************************************* --------------------------------------------------------- Relate * Authors: Leo Speidel, Marie Forest, Sinan Shi, Simon Myers. * Doc: https://myersgroup.github.io/relate --------------------------------------------------------- --------------------------------------------------------- Using: Results/prep.chr2.haps.gz Results/prep.chr2.sample.gz relate_data/genetic_map_chr2_combined_b37.txt with mu = 1.25e-08 and 2Ne = 30000. --------------------------------------------------------- --------------------------------------------------------- Parsing data.. Warning: Will use min 0.00035GB of hard disc. CPU Time spent: 0.682457s; Max Memory usage: 23Mb. --------------------------------------------------------- --------------------------------------------------------- Read 120 haplotypes with 67935 SNPs per haplotype. Expected minimum memory usage: 0.61Gb. --------------------------------------------------------- --------------------------------------------------------- Starting chunk 0 of 0. --------------------------------------------------------- --------------------------------------------------------- Painting sequences... CPU Time spent: 1.655027s; Max Memory usage: 23Mb. --------------------------------------------------------- --------------------------------------------------------- Estimating topologies of AncesTrees in sections 0-0... CPU Time spent: 5.264306s; Max Memory usage: 8.2e+02Mb. --------------------------------------------------------- --------------------------------------------------------- Propagating mutations across AncesTrees... CPU Time spent: 5.952430s; Max Memory usage: 8.2e+02Mb. --------------------------------------------------------- --------------------------------------------------------- Inferring branch lengths of AncesTrees in sections 0-0... CPU Time spent: 46.492151s; Max Memory usage: 8.2e+02Mb. --------------------------------------------------------- --------------------------------------------------------- Combining AncesTrees in Sections... CPU Time spent: 46.787977s; Max Memory usage: 8.2e+02Mb. --------------------------------------------------------- --------------------------------------------------------- Finalizing... Number of not mapping SNPs: 529 Number of flipped SNPs : 274 CPU Time spent: 47.764615s; Max Memory usage: 8.2e+02Mb. --------------------------------------------------------- --------------------------------------------------------- Done. --------------------------------------------------------- *********************************************************
mv chr2_relate.* ./Results/ #moving output into Results folder
- Q3: How many SNPs are left per haplotype?
Estimation of population size¶
The lengthiest process is this step, in which population size is estimated, and the population size is re-estimates branch lengths. This takes up to 20 minutes. While you wait, open the notebook 07b_interactive_exercise and try to answer to the questions trying out the interactive interface.
relate/scripts/EstimatePopulationSize/EstimatePopulationSize.sh -i Results/chr2_relate \
-m 1.25e-8 --poplabels relate_data/60_inds.txt \
-o Results/popsize --threshold 0
********************************
Parameters passed to script:
input = Results/chr2_relate
poplabels = relate_data/60_inds.txt
mu = 1.25e-8
years_per_gen = 28
output = Results/popsize
num_iter = 10
threshold = 0
Maximum number of threads: 1
********************************
---------------------------------------------------------
Extracting dist file from Results/chr2_relate.mut ...
CPU Time spent: 0.083929s; Max Memory usage: 58.744Mb.
---------------------------------------------------------
---------------------------------------------------------
Removing trees with few mutations from Results/chr2_relate.mut ...
CPU Time spent: 2.502428s; Max Memory usage: 282.068Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.765854s; Max Memory usage: 59.904Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.845644s; Max Memory usage: 168.776Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.749195s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 68.056845s; Max Memory usage: 168.892Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.745532s; Max Memory usage: 59.904Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 66.573337s; Max Memory usage: 168.888Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.756833s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.010154s; Max Memory usage: 168.892Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.750237s; Max Memory usage: 59.904Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.448596s; Max Memory usage: 168.888Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.740143s; Max Memory usage: 59.904Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.362031s; Max Memory usage: 168.892Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.741483s; Max Memory usage: 59.904Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 66.451561s; Max Memory usage: 168.892Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.750135s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.161951s; Max Memory usage: 168.892Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.789958s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.409804s; Max Memory usage: 168.724Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.815423s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Sampling branch lengths for Results/popsize ...
[100%]
CPU Time spent: 67.701972s; Max Memory usage: 168.656Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rates for Results/popsize...
[100%]
CPU Time spent: 0.771737s; Max Memory usage: 59.972Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating coalescence rate for Results/popsize ...
CPU Time spent: 2.466906s; Max Memory usage: 119.632Mb.
---------------------------------------------------------
---------------------------------------------------------
Finalizing coalescence rate...
|
6.90e+04|**
|
| **
| **** **
| ** ** **
| ******** ** ** **** **
| ** ** **
| ** **
| ** **
| ** **
1.51e+03| **
|
-----------------------------------------------------------
3.57e+01 3.57e+05
generations ago
CPU Time spent: 2.488972s; Max Memory usage: 119.632Mb.
---------------------------------------------------------
---------------------------------------------------------
Calculating average mutation rate for Results/popsize ...
|
1.81e-08| **
|** ******
| ****
| **
| **
| ****
| **** **
| ************ **
|
| ****
1.27e-08| ********
|
-----------------------------------------------------------
3.57e+01 3.57e+05
generations ago
CPU Time spent: 0.846613s; Max Memory usage: 118.66Mb.
---------------------------------------------------------
---------------------------------------------------------
Reinferring branch lengths for Results/chr2_relate ...
[100%]
CPU Time spent: 62.879230s; Max Memory usage: 110.744Mb.
---------------------------------------------------------
---------------------------------------------------------
Finalizing coalescence rate...
CPU Time spent: 0.006047s; Max Memory usage: 2.616Mb.
---------------------------------------------------------
Warning message:
Transformation introduced infinite values in continuous x-axis
null device
1
- Q4: At the end, Relate outputs estimated mutation rate and coalescence times along the region - can this tell us anything?
Selection¶
With the coalescence rates estimated, it is possible to detect selection.
relate/scripts/DetectSelection/DetectSelection.sh -i Results/popsize -m 1.25e-8 \
--poplabels relate_data/60_inds.txt \
-o Results/selection_relate
******************************** Parameters passed to script: input = Results/popsize mu = 1.25e-8 years_per_gen = 28 output = Results/selection_relate ******************************** --------------------------------------------------------- Calculating frequency through time for Results/popsize. CPU Time spent: 1.368211s; Max Memory usage: 117.668Mb. --------------------------------------------------------- --------------------------------------------------------- Calculating evidence of selection for Results/selection_relate. CPU Time spent: 2.730782s; Max Memory usage: 1.16Mb. ---------------------------------------------------------
At this point, we are detecting selection based on three distinct populations.
Q5: Consider whether the underlying assumptions for this make sense.
To find the sites with the strongest selection, we will sort based on col 35 (which is the one with the log10 p-value for selection). The first command is used for sorting. You will see the positions in the first column of the output.
In the second command we sort again, but isolate the first column (genomic position) and column 35 (log10 p-value for selection), and then keep only the first 10 rows (excluding the names pos and when_mutation_has_freq2 in the first row). Change the number 11 in the command sed -n '2,11'p to choose how many lines to print.
sort -k35r Results/selection_relate.sele | head -n 10
pos rs_id 3571428.500000 357142.937500 257030.656250 184981.281250 133128.375000 95810.585938 68953.507812 49624.847656 35714.292969 25703.064453 18498.126953 13312.836914 9581.057617 6895.350098 4962.484863 3571.429199 2570.306396 1849.812622 1331.283691 958.105774 689.534973 496.248444 357.142914 257.030640 184.981262 133.128357 95.810577 68.953499 49.624844 35.714287 0.000000 when_DAF_is_half when_mutation_has_freq2 143026077 rs59280189 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -6.09684 -5.36534 -4.28703 -3.68305 -3.25563 -2.23481 -1.05641 -1.09153 -0.66056 -0.448427 -0.309981 -0.0787622 0 0 0 0 0 -1.80559 -6.09684 136608646 rs4988235 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -5.01126 -5.96369 -5.76504 -5.91704 -4.49533 -1.23787 0 0 0 0 -6.35246 -5.47079 136616754 rs182549 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -5.01126 -5.96369 -5.76504 -5.91704 -4.49533 -1.23787 0 0 0 0 -6.35246 -5.47079 131110097 rs6750645 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -4.12253 -3.61525 -2.12713 -1.15708 -1.03286 -0.692444 -1.17085 -1.04607 -0.773465 -0.381002 -0.0568152 0 0 0 -1.28724 -5.46694 141653595 rs12469352 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -4.04941 -1.95932 -1.53461 -1.60475 -2.50727 -1.37845 -0.704867 -0.484479 0 0 0 0 -1.83952 -5.23884 130500081 rs13017715 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -3.935 -2.65248 -1.38281 -1.48468 -0.936435 -0.526179 -0.558078 -0.265231 -0.0159609 0 0 0 0 0 -2.22373 -5.23617 136169799 rs6742013 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -5.82528 -6.7453 -5.40901 -2.27544 -1.81653 -2.41799 -1.72666 -1.78017 -0.887542 -0.218208 0 0 0 -1.91098 -5.21295 130498804 rs10928929 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -4.5755 -5.78962 -4.54734 -3.94768 -3.19114 -3.34317 -1.45072 -0.908191 -0.626947 -0.182633 -0.0296485 -0.0107089 -0.0107089 0 0 0 -2.99375 -5.18567 130499359 rs4233597 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 -4.5755 -5.78962 -4.54734 -3.94768 -3.19114 -3.34317 -1.45072 -0.908191 -0.626947 -0.182633 -0.0296485 -0.0107089 -0.0107089 0 0 0 -2.99375 -5.18567 sort: write failed: 'standard output': Broken pipe sort: write error
sort -k35r Results/selection_relate.sele | cut -f1,35 -d' ' | sed -n '2,11'p
143026077 -6.09684 136608646 -5.47079 136616754 -5.47079 131110097 -5.46694 141653595 -5.23884 130500081 -5.23617 136169799 -5.21295 130498804 -5.18567 130499359 -5.18567 139393537 -5.03088
Now we know position and p-value for the highest positive selections. Try plotting some of the sites with the following command. Use -o to determine the output name of the plot, and bp_of_interest for the genomic position. Below is an example for position 143026077. We change the output name according to position, so that we will not overwrite the plots. The output is a pdf plot with name Results/tree_POSITION.pdf
POSITION=143026077
relate/scripts/TreeView/TreeView.sh --haps Results/chr2.haps --sample Results/chr2.sample \
--anc Results/popsize.anc --mut Results/popsize.mut \
--poplabels relate_data/60_inds.txt --years_per_gen 28 \
-o Results/tree_$POSITION --bp_of_interest $POSITION
********************************
Parameters passed to script:
haps = Results/chr2.haps
sample = Results/chr2.sample
poplabels = relate_data/60_inds.txt
anc = Results/popsize.anc
mut = Results/popsize.mut
years_per_gen = 28
bp_of_interest = 143026077
output = Results/tree_143026077.pdf
********************************
Loading required package: dplyr
Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Loading required package: ggplot2
Loading required package: cowplot
Installing package into ‘/usr/local/spark-3.2.1-bin-hadoop3.2/R/lib’
(as ‘lib’ is unspecified)
trying URL 'http://cran.us.r-project.org/src/contrib/cowplot_1.1.1.tar.gz'
Content type 'application/x-gzip' length 1353271 bytes (1.3 MB)
==================================================
downloaded 1.3 MB
* installing *source* package ‘cowplot’ ...
** package ‘cowplot’ successfully unpacked and MD5 sums checked
** using staged installation
** R
** inst
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
*** copying figures
** building package indices
** installing vignettes
** testing if installed package can be loaded from temporary location
** testing if installed package can be loaded from final location
** testing if installed package keeps a record of temporary installation path
* DONE (cowplot)
The downloaded source packages are in
‘/tmp/RtmpSu166h/downloaded_packages’
Loading required package: cowplot
Warning message:
In library(package, lib.loc = lib.loc, character.only = TRUE, logical.return = TRUE, :
there is no package called ‘cowplot’
---------------------------------------------------------
Get tree at BP 143026077...
CPU Time spent: 0.686335s; Max Memory usage: 117.704Mb.
---------------------------------------------------------
---------------------------------------------------------
Get list of mutations that map to tree at BP 143026077...
CPU Time spent: 1.072413s; Max Memory usage: 206.456Mb.
---------------------------------------------------------
Warning message:
`guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
null device
1
Q6: Is it the same populations in which the sweep seem to occur?
Q7: The various populations are generally not fully split. Consider why Relate can end up inferring this.
Q8: For a site which seems promising, figure out what the nearby genes code for.
Exercise: perform the analysis for a single population¶
In general, selection scans of this kind will perform better if it is for a single population. Try rerunning the analysis with pure brits. Copy and paste code from the previous commands and adapt them to the new data
Necessary files for this are as well in the relate_data folder, and are
- relate_data/50_GBR_inds.txt
- relate_data/chr2_GBR_phased.vcf.gz
which substitute the files 60_inds.txt and chr2_130_145_phased.vcf.gz. The rest of the files needed are still the same as before.
When you are done, answer to the following questions:
- Q9: Compared to the results in Q6, do you get different or similar results?
- Q10: Try plotting some of the same sites, and see whether the trees (when considering GBR) are similar.