Quality Control: Strand and mapping correction, anchoring and population covariates
Important notes for this notebook
In this section, we will explore how to match strands and reference mapping to merge HapMap and 1000GP data. Then we will use population information from one dataset to decide if there are ethnic outliers in the other. Finally, we generate population covariates useful for association testing.
Learning outcomes
Evaluate when and how to apply strand and mapping correction
Discuss confounding factors that lead to spurious associations
Identify population substructure and filter out outliers
Correct for population stratification
How to make this notebook work
In this notebook, we will both use R and bash command line programming languages. Remember to change the kernel whenever you transition from one language to the other (Kernel --> Change Kernel) indicated by the languages’ images.
We will first run Bash commands.
Choose the Bash kernel
Merging, anchoring and making covariates
Population stratification presents a significant source of systematic bias in GWAS, arising when subpopulations exhibit systematic differences in allele frequencies. Research indicates that even subtle degrees of population stratification can exist within a single ethnic population (Abdellaoui et al. 2013). Thus, testing and controlling for the presence of population stratification is an essential QC step.
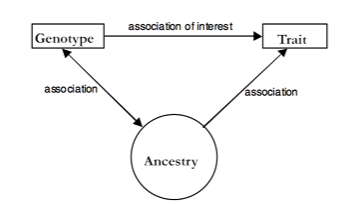
The population structure (or in other words, the ancestral relationship of the populations) is a so-called confounding factor. This means that it affects both the dependent and independent variables, as shown in the figure below, where both the genotype and traits are influenced by population structure (e.g., the distribution of north and south European individuals in the PCA space and the height of those individuals).
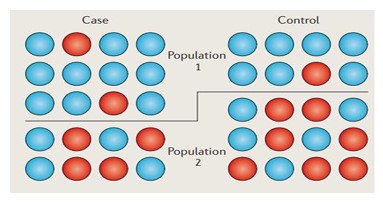
Why is a bias introduced? Population structure can influence allele frequencies and produce false positives/negatives when doing association testing. Graphically, consider the example in the figure below. Case and control for population 1 and 2 have specific MAFs. Removing the barrier between populations will change MAFs drastically for the two confitions.
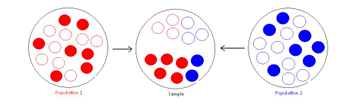
The same problem arises in population studies without Case-control categories. Imagine having a population of randomly sampled individuals, each from a different ethnicity (the blue and red minor alleles in the example below). The final group of individuals will have a different proportion of MAFs depending on the sampling of various ethnicities.
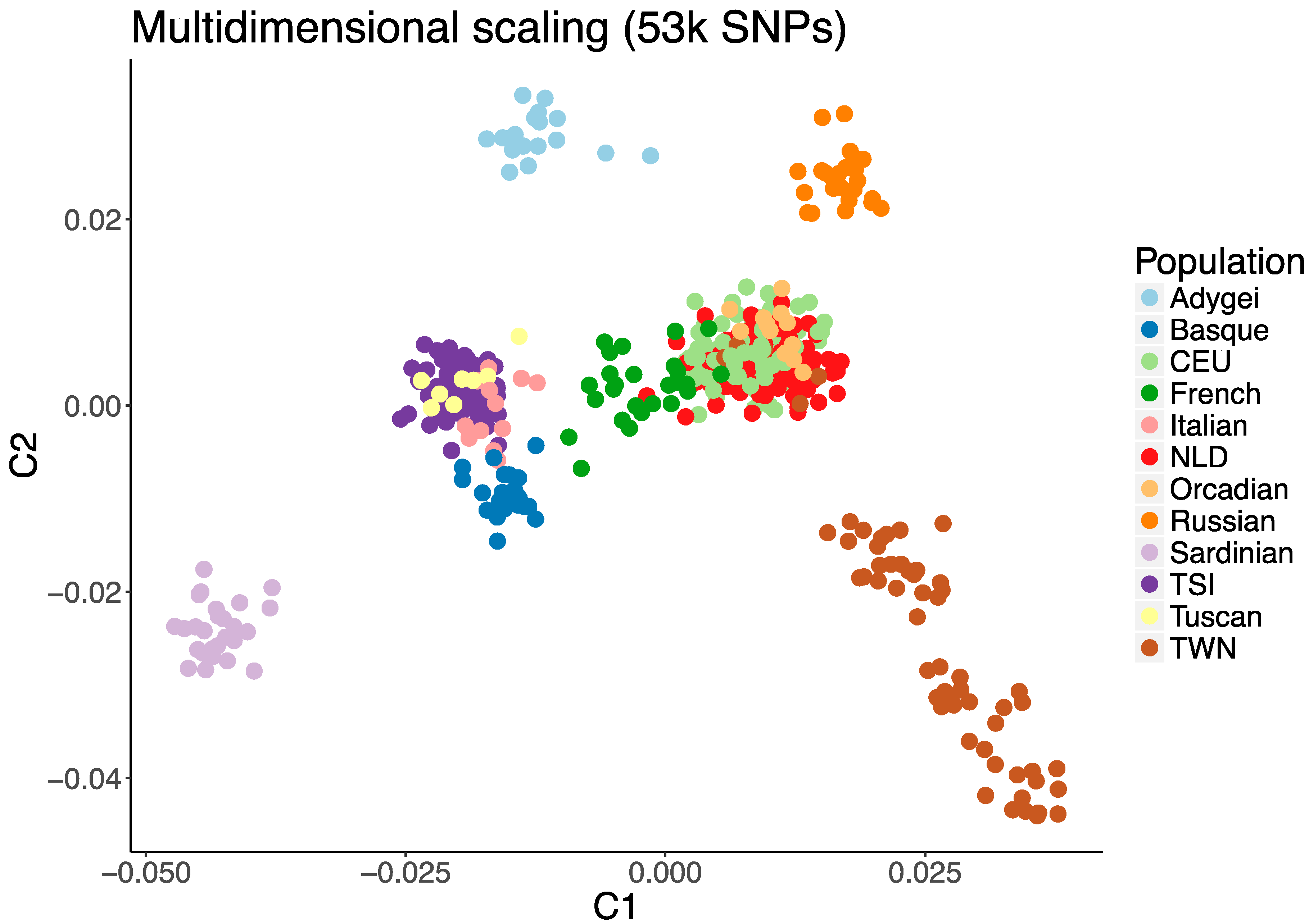
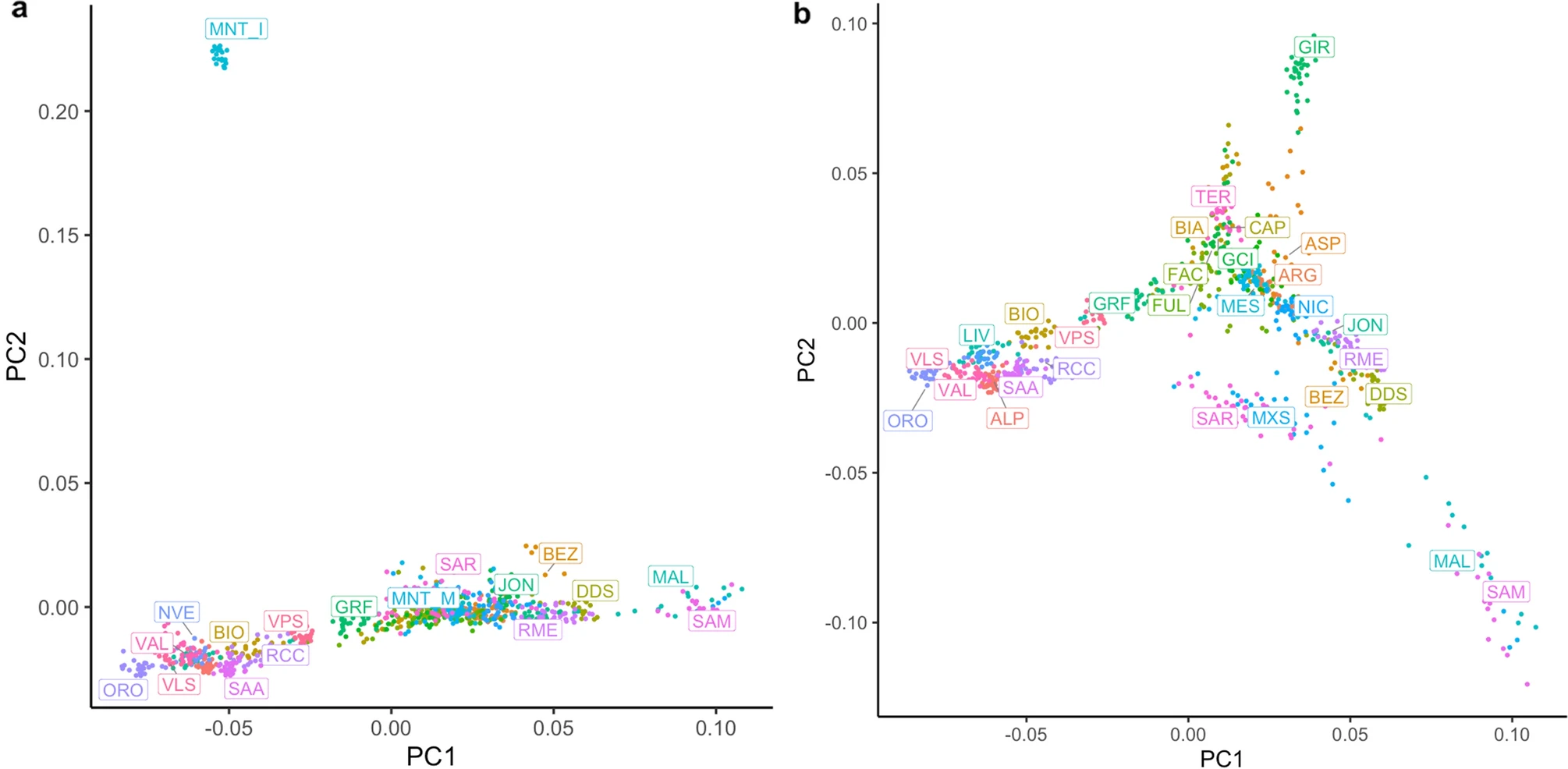
There are several methods to correct for population stratification Alkes L. Price et al. (2006). Here, we illustrate a method integrated into PLINK: the multidimensional scaling (MDS) approach. MDS calculates the genome-wide average proportion of shared alleles between any pair of individuals to generate quantitative indices (components) of the genetic variation for each individual. The individual component scores can be visualized to identify groups of genetically similar individuals. For instance, in a genetic study including subjects from Asia and Europe, MDS analysis would reveal that Asians are genetically more similar to each other than to Europeans and Africans. The figure below shows another example of MDS using HapMap, Genome diversity project, and authors’ data:
Example of MDS plot of the first two MDS components scores on integrated datasets in Somers et al. (2017). Here TWN, a past isolated population in the Netherlands, is separated from the other individuals from the same country (NDL). Sardinians, notoriously different from continental Italians, are also grouped far away from them. Central European populations colocalize as expected.
Anchoring
To investigate which individuals the generated component scores deviate from in the target population, plotting the scores of the dataset under investigation and a population of known ethnic structure (e.g., HapMap/1KG data) is helpful: this step is called anchoring(Rietveld et al. 2013). This enables the researcher to obtain ethnic information on their data and to determine possible ethnic outliers. For example, in the figure above, if TSI (Tuscans from Italy) is the anchor population, one can hypothesize that the yellow dots might be ethnically similar (as in the example).
Outlier removal
Outliers identified based on MDS analysis should be excluded from further analyses. Following their removal, a new MDS analysis must be conducted, and its primary components are utilized as covariates in association tests to correct for any residual population stratification within the population. The number of components to include depends on the population structure and sample size (usually 10-20).
The MDS from Cortellari et al. (2021) shows a distinct goat population outlier. The second axis is dominated by this outlier, obscuring structure in the other populations. Removing the outlier reveals a clearer structure among the remaining populations.
Analysis
We aim to merge the HapMap and 1000GP datasets, using 1000GP Phase I as the anchor for HapMap. Our goal is to check if we can identify the ethnicity of the HapMap data based on the ethnicities in the 1000GP dataset. There are several steps to ensure compatibility between the datasets, so stay with us!
1000GP data download
Here are some commands to download and convert the 1000GP data for GWAS analysis. You don’t need to run them, as we’ve already processed the data.
1000 Genomes Project - Phase I: genetic information for 629 individuals from various ethnic groups (>60GB). Phase III is now available, and we recommend using it for research purposes.
The 1000 Genomes data downloaded above is rather large so the commands are not executable and are shown for reference only. To save time, we’ve provided the .bed, .bimand .fam files in the Data folder.
Let’s unzip the files and see how many samples we have.
unzip-o Data/1000genomes.zip -d Results/GWAS4# count lines in fam wc-l Results/GWAS4/1000genomes.genotypesA.fam
1 rs112750067 0 10327 C T
1 . 0 11508 A G
1 . 0 12783 G A
1 . 0 13116 G T
1 . 0 14933 A G
One should note that the file 1000genomes.genotypes.bim contains SNPs without an rs-identifier (or Reference SNP cluster ID). The missing rs-identifiers (noted as .) are not a problem for this tutorial. However, for good practice, we will assign unique identifiers to the SNPs (using available information):
Now, let’s visualize the data to check the SNP names assigned. These are derived from the format @:#[b37]\$1,\$2 in the command above, which PLINK interprets as chromosome:locus[b37]Allele1,Allele2:
# Show changes on the bim filecat Results/GWAS4/1000genomes.genotypesA_no_missing_IDs.bim |head-5
1 rs112750067 0 10327 C T
1 1:11508[b37]A,G 0 11508 A G
1 1:12783[b37]A,G 0 12783 G A
1 1:13116[b37]G,T 0 13116 G T
1 1:14933[b37]A,G 0 14933 A G
Pretty neat, right?
QC on 1000GP data
As we covered in the GWAS3 notebook, it’s important to account for missingness, sex discrepancies, and minor allele frequency. We’ll apply standard QC thresholds to the 1000 GP data before merging it with HapMap data.
N.B: Ensure that the datasets you want to merge share the same genomic build! Otherwise, you’ll need to include a liftover step.
We want to only consider SNPs that both datasets have in common. First, extract SNP names from the HapMap data and filter the 1000GP data to include only matching SNPs.
#Print out SNPs from the HapMap dataawk'{print$2}' Results/GWAS4/HapMap_3_r3_9.bim > Results/GWAS4/HapMap_SNPs.txt#Extract the HapMap SNPs from the 1000GP dataplink--bfile Results/GWAS4/1kG_MDS3 \--extract Results/GWAS4/HapMap_SNPs.txt \--make-bed\--out Results/GWAS4/1kG_MDS4
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/1kG_MDS4.log.
Options in effect:
--bfile Results/GWAS4/1kG_MDS3
--extract Results/GWAS4/HapMap_SNPs.txt
--make-bed
--out Results/GWAS4/1kG_MDS4
385567 MB RAM detected; reserving 192783 MB for main workspace.
2232052 variants loaded from .bim file.
37 people (0 males, 0 females, 37 ambiguous) loaded from .fam.
Ambiguous sex IDs written to Results/GWAS4/1kG_MDS4.nosex .
--extract: 376380 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 37 founders and 0 nonfounders present.
Calculating allele frequencies... done.
376380 variants and 37 people pass filters and QC.
Note: No phenotypes present.
--make-bed to Results/GWAS4/1kG_MDS4.bed + Results/GWAS4/1kG_MDS4.bim +
Results/GWAS4/1kG_MDS4.fam ... done.
This is how part of the list of SNP names looks like:
Now we take the variants from the reduced 1000GP data, and go the other way around. We extract 1000GP variants from the HapMap data. In other words, the two extraction passages will intersect the SNPs. Below is the code to use the SNPs of the 1000GP data to reduce the HapMap data.
#Print out SNPs from the HapMap dataawk'{print$2}' Results/GWAS4/1kG_MDS4.bim > Results/GWAS4/1kG_MDS4_SNPs.txt#Extract the HapMap SNPs from the 1000GP dataplink--bfile Results/GWAS4/HapMap_3_r3_9 \--extract Results/GWAS4/1kG_MDS4_SNPs.txt \--make-bed\--out Results/GWAS4/HapMap_MDS
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/HapMap_MDS.log.
Options in effect:
--bfile Results/GWAS4/HapMap_3_r3_9
--extract Results/GWAS4/1kG_MDS4_SNPs.txt
--make-bed
--out Results/GWAS4/HapMap_MDS
385567 MB RAM detected; reserving 192783 MB for main workspace.
1073226 variants loaded from .bim file.
109 people (55 males, 54 females) loaded from .fam.
109 phenotype values loaded from .fam.
--extract: 376380 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 109 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.998025.
376380 variants and 109 people pass filters and QC.
Among remaining phenotypes, 54 are cases and 55 are controls.
--make-bed to Results/GWAS4/HapMap_MDS.bed + Results/GWAS4/HapMap_MDS.bim +
Results/GWAS4/HapMap_MDS.fam ... done.
Stop - Read - Solve
Look at the two outputs below a bit more carefully. The names are all matching. Is there any other problem?
head Results/GWAS4/HapMap_MDS.bim
1 rs3131969 0 744045 A G
1 rs12562034 0 758311 A G
1 rs4970383 0 828418 A C
1 rs4475691 0 836671 T C
1 rs1806509 0 843817 C A
1 rs28576697 0 860508 C T
1 rs3748595 0 877423 A C
1 rs13303118 0 908247 G T
1 rs1891910 0 922320 A G
1 rs3128097 0 970323 G A
head Results/GWAS4/1kG_MDS4.bim
1 rs3131969 0 754182 A G
1 rs12562034 0 768448 A G
1 rs4970383 0 838555 A C
1 rs4475691 0 846808 T C
1 rs1806509 0 853954 A C
1 rs28576697 0 870645 C T
1 rs3748595 0 887560 A C
1 rs13303118 0 918384 T G
1 rs1891910 0 932457 A G
1 rs3128097 0 980460 G A
Build matching
Genomic data is based on a reference genome, and our datasets use different human reference versions. Since the reference genome improves over time, SNP positions may differ between datasets from different versions.
We extract SNP names and positions from the HapMap data and align the 1000GP data to match these SNPs using the --update-map option in PLINK.
#Extract the HapMap variant coordinatesawk'{print$2,$4}' Results/GWAS4/HapMap_MDS.bim > Results/GWAS4/buildhapmap.txt
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/1kG_MDS5.log.
Options in effect:
--bfile Results/GWAS4/1kG_MDS4
--make-bed
--out Results/GWAS4/1kG_MDS5
--update-map Results/GWAS4/buildhapmap.txt
385567 MB RAM detected; reserving 192783 MB for main workspace.
376380 variants loaded from .bim file.
37 people (0 males, 0 females, 37 ambiguous) loaded from .fam.
Ambiguous sex IDs written to Results/GWAS4/1kG_MDS5.nosex .
--update-map: 376380 values updated.
Warning: Base-pair positions are now unsorted!
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 37 founders and 0 nonfounders present.
Calculating allele frequencies... done.
376380 variants and 37 people pass filters and QC.
Note: No phenotypes present.
--make-bed to Results/GWAS4/1kG_MDS5.bed + Results/GWAS4/1kG_MDS5.bim +
Results/GWAS4/1kG_MDS5.fam ... done.
Merging datasets and performing MDS
Before merging the HapMap and 1000 Genomes datasets, we ensure compatibility through 3 steps:
Verify the reference genome is compatible in both datasets.
Align SNP orientations (strand) across datasets.
Remove SNPs that still differ after these steps.
The next steps are technical but ensure the datasets correspond correctly.
1. We’ve matched SNP positions, but we also need to ensure the reference alleles align. Remember that most PLINK analyses consider the A1 allele (typically the minor allele) as the reference allele, which is logical when dealing exclusively with biallelic variants.
Below, we generate a list of SNPs ID and ‘reference alleles’ (corresponding to A1, column 5 of the .bim file) from 1000GP.
#Extract variant coordinates and reference alleles from 1000GP dataawk'{print$2,$5}' Results/GWAS4/1kG_MDS5.bim > Results/GWAS4/1kg_ref-list.txt
How the list looks like:
head-5 Results/GWAS4/1kg_ref-list.txt
rs3131969 A
rs12562034 A
rs4970383 A
rs4475691 T
rs1806509 A
Then, we assign them to the HapMap data --ref-allele option. Note a lot fo warnings in A1 allele assignment. This is usually because you have a strand issue, where the data uses a complement
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/HapMap-adj.log.
Options in effect:
--a1-allele Results/GWAS4/1kg_ref-list.txt
--bfile Results/GWAS4/HapMap_MDS
--make-bed
--out Results/GWAS4/HapMap-adj
385567 MB RAM detected; reserving 192783 MB for main workspace.
376380 variants loaded from .bim file.
109 people (55 males, 54 females) loaded from .fam.
109 phenotype values loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 109 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.998025.
Warning: Impossible A1 allele assignment for variant rs11488462.
Warning: Impossible A1 allele assignment for variant rs28635343.
Warning: Impossible A1 allele assignment for variant rs28456011.
Warning: Impossible A1 allele assignment for variant rs28487995.
Warning: Impossible A1 allele assignment for variant rs760925.
Warning: Impossible A1 allele assignment for variant rs2234167.
Warning: Impossible A1 allele assignment for variant rs35383955.
Warning: Impossible A1 allele assignment for variant rs34283457.
Warning: Impossible A1 allele assignment for variant rs35357981.
Warning: Impossible A1 allele assignment for variant rs35025175.
Warning: Impossible A1 allele assignment for variant rs35974482.
Warning: Impossible A1 allele assignment for variant rs34835780.
Warning: Impossible A1 allele assignment for variant rs10753374.
Warning: Impossible A1 allele assignment for variant rs11260657.
Warning: Impossible A1 allele assignment for variant rs4661727.
Warning: Impossible A1 allele assignment for variant rs35614701.
Warning: Impossible A1 allele assignment for variant rs34131388.
Warning: Impossible A1 allele assignment for variant rs35260034.
Warning: Impossible A1 allele assignment for variant rs34201264.
Warning: Impossible A1 allele assignment for variant rs34734086.
Warning: Impossible A1 allele assignment for variant rs35593799.
Warning: Impossible A1 allele assignment for variant rs35047308.
Warning: Impossible A1 allele assignment for variant rs36053581.
Warning: Impossible A1 allele assignment for variant rs34405972.
Warning: Impossible A1 allele assignment for variant rs35294772.
Warning: Impossible A1 allele assignment for variant rs12118489.
Warning: Impossible A1 allele assignment for variant rs1023098.
Warning: Impossible A1 allele assignment for variant rs209603.
Warning: Impossible A1 allele assignment for variant rs10524.
Warning: Impossible A1 allele assignment for variant rs697594.
Warning: Impossible A1 allele assignment for variant rs1889603.
Warning: Impossible A1 allele assignment for variant rs12145114.
Warning: Impossible A1 allele assignment for variant rs2391154.
Warning: Impossible A1 allele assignment for variant rs17123142.
Warning: Impossible A1 allele assignment for variant rs12125356.
Warning: Impossible A1 allele assignment for variant rs3765501.
Warning: Impossible A1 allele assignment for variant rs1011297.
Warning: Impossible A1 allele assignment for variant rs34444588.
Warning: Impossible A1 allele assignment for variant rs509587.
Warning: Impossible A1 allele assignment for variant rs17701857.
Warning: Impossible A1 allele assignment for variant rs3795743.
Warning: Impossible A1 allele assignment for variant rs34158997.
Warning: Impossible A1 allele assignment for variant rs35808016.
Warning: Impossible A1 allele assignment for variant rs34187970.
Warning: Impossible A1 allele assignment for variant rs3820624.
Warning: Impossible A1 allele assignment for variant rs2897454.
Warning: Impossible A1 allele assignment for variant rs1127975.
Warning: Impossible A1 allele assignment for variant rs35729322.
Warning: Impossible A1 allele assignment for variant rs35565142.
Warning: Impossible A1 allele assignment for variant rs3007405.
Warning: Impossible A1 allele assignment for variant rs11553746.
Warning: Impossible A1 allele assignment for variant rs1384491.
Warning: Impossible A1 allele assignment for variant rs17045393.
Warning: Impossible A1 allele assignment for variant rs6545238.
Warning: Impossible A1 allele assignment for variant rs17792521.
Warning: Impossible A1 allele assignment for variant rs989586.
Warning: Impossible A1 allele assignment for variant rs7558635.
Warning: Impossible A1 allele assignment for variant rs1139829.
Warning: Impossible A1 allele assignment for variant rs35061433.
Warning: Impossible A1 allele assignment for variant rs838069.
Warning: Impossible A1 allele assignment for variant rs967381.
Warning: Impossible A1 allele assignment for variant rs2138486.
Warning: Impossible A1 allele assignment for variant rs13393016.
Warning: Impossible A1 allele assignment for variant rs17180544.
Warning: Impossible A1 allele assignment for variant rs13417895.
Warning: Impossible A1 allele assignment for variant rs12467878.
Warning: Impossible A1 allele assignment for variant rs6726184.
Warning: Impossible A1 allele assignment for variant rs4973697.
Warning: Impossible A1 allele assignment for variant rs6605267.
Warning: Impossible A1 allele assignment for variant rs7421596.
Warning: Impossible A1 allele assignment for variant rs1689581.
Warning: Impossible A1 allele assignment for variant rs711730.
Warning: Impossible A1 allele assignment for variant rs7652667.
Warning: Impossible A1 allele assignment for variant rs9864701.
Warning: Impossible A1 allele assignment for variant rs1522553.
Warning: Impossible A1 allele assignment for variant rs17280613.
Warning: Impossible A1 allele assignment for variant rs277646.
Warning: Impossible A1 allele assignment for variant rs1259321.
Warning: Impossible A1 allele assignment for variant rs6765489.
Warning: Impossible A1 allele assignment for variant rs1113277.
Warning: Impossible A1 allele assignment for variant rs9880098.
Warning: Impossible A1 allele assignment for variant rs7611483.
Warning: Impossible A1 allele assignment for variant rs10936388.
Warning: Impossible A1 allele assignment for variant rs11546878.
Warning: Impossible A1 allele assignment for variant rs4686566.
Warning: Impossible A1 allele assignment for variant rs884309.
Warning: Impossible A1 allele assignment for variant rs3806620.
Warning: Impossible A1 allele assignment for variant rs6801044.
Warning: Impossible A1 allele assignment for variant rs4677689.
Warning: Impossible A1 allele assignment for variant rs4677695.
Warning: Impossible A1 allele assignment for variant rs9820715.
Warning: Impossible A1 allele assignment for variant rs35840880.
Warning: Impossible A1 allele assignment for variant rs6847677.
Warning: Impossible A1 allele assignment for variant rs16854250.
Warning: Impossible A1 allele assignment for variant rs12646999.
Warning: Impossible A1 allele assignment for variant rs13128530.
Warning: Impossible A1 allele assignment for variant rs2949614.
Warning: Impossible A1 allele assignment for variant rs17007758.
Warning: Impossible A1 allele assignment for variant rs10006274.
Warning: Impossible A1 allele assignment for variant rs4691482.
Warning: Impossible A1 allele assignment for variant rs1215429.
Warning: Impossible A1 allele assignment for variant rs1395093.
Warning: Impossible A1 allele assignment for variant rs2972819.
Warning: Impossible A1 allele assignment for variant rs199361.
Warning: Impossible A1 allele assignment for variant rs4403186.
Warning: Impossible A1 allele assignment for variant rs1574436.
Warning: Impossible A1 allele assignment for variant rs1048944.
Warning: Impossible A1 allele assignment for variant rs4704197.
Warning: Impossible A1 allele assignment for variant rs173545.
Warning: Impossible A1 allele assignment for variant rs2060424.
Warning: Impossible A1 allele assignment for variant rs449359.
Warning: Impossible A1 allele assignment for variant rs11746705.
Warning: Impossible A1 allele assignment for variant rs7448017.
Warning: Impossible A1 allele assignment for variant rs1465686.
Warning: Impossible A1 allele assignment for variant rs13180237.
Warning: Impossible A1 allele assignment for variant rs17776554.
Warning: Impossible A1 allele assignment for variant rs266000.
Warning: Impossible A1 allele assignment for variant rs4868581.
Warning: Impossible A1 allele assignment for variant rs4710897.
Warning: Impossible A1 allele assignment for variant rs9461653.
Warning: Impossible A1 allele assignment for variant rs1129765.
Warning: Impossible A1 allele assignment for variant rs1694112.
Warning: Impossible A1 allele assignment for variant rs12207915.
Warning: Impossible A1 allele assignment for variant rs1383266.
Warning: Impossible A1 allele assignment for variant rs4565302.
Warning: Impossible A1 allele assignment for variant rs970392.
Warning: Impossible A1 allele assignment for variant rs17183312.
Warning: Impossible A1 allele assignment for variant rs2075967.
Warning: Impossible A1 allele assignment for variant rs3757212.
Warning: Impossible A1 allele assignment for variant rs573684.
Warning: Impossible A1 allele assignment for variant rs9373596.
Warning: Impossible A1 allele assignment for variant rs3924019.
Warning: Impossible A1 allele assignment for variant rs9719226.
Warning: Impossible A1 allele assignment for variant rs2961253.
Warning: Impossible A1 allele assignment for variant rs2428430.
Warning: Impossible A1 allele assignment for variant rs4870666.
Warning: Impossible A1 allele assignment for variant rs1043987.
Warning: Impossible A1 allele assignment for variant rs2068338.
Warning: Impossible A1 allele assignment for variant rs2283017.
Warning: Impossible A1 allele assignment for variant rs2854541.
Warning: Impossible A1 allele assignment for variant rs361489.
Warning: Impossible A1 allele assignment for variant rs2855882.
Warning: Impossible A1 allele assignment for variant rs2734060.
Warning: Impossible A1 allele assignment for variant rs2244520.
Warning: Impossible A1 allele assignment for variant rs1573618.
Warning: Impossible A1 allele assignment for variant rs2855914.
Warning: Impossible A1 allele assignment for variant rs2734112.
Warning: Impossible A1 allele assignment for variant rs2367191.
Warning: Impossible A1 allele assignment for variant rs2855920.
Warning: Impossible A1 allele assignment for variant rs2855929.
Warning: Impossible A1 allele assignment for variant rs6961143.
Warning: Impossible A1 allele assignment for variant rs17231.
Warning: Impossible A1 allele assignment for variant rs6979421.
Warning: Impossible A1 allele assignment for variant rs17163237.
Warning: Impossible A1 allele assignment for variant rs1008660.
Warning: Impossible A1 allele assignment for variant rs17250.
Warning: Impossible A1 allele assignment for variant rs2156940.
Warning: Impossible A1 allele assignment for variant rs11768792.
Warning: Impossible A1 allele assignment for variant rs17277.
Warning: Impossible A1 allele assignment for variant rs17279.
Warning: Impossible A1 allele assignment for variant rs2734171.
Warning: Impossible A1 allele assignment for variant rs6979469.
Warning: Impossible A1 allele assignment for variant rs2156956.
Warning: Impossible A1 allele assignment for variant rs6943682.
Warning: Impossible A1 allele assignment for variant rs926044.
Warning: Impossible A1 allele assignment for variant rs6971657.
Warning: Impossible A1 allele assignment for variant rs6942393.
Warning: Impossible A1 allele assignment for variant rs1134309.
Warning: Impossible A1 allele assignment for variant rs17835147.
Warning: Impossible A1 allele assignment for variant rs1114856.
Warning: Impossible A1 allele assignment for variant rs10088098.
Warning: Impossible A1 allele assignment for variant rs11780139.
Warning: Impossible A1 allele assignment for variant rs39767.
Warning: Impossible A1 allele assignment for variant rs1027623.
Warning: Impossible A1 allele assignment for variant rs1545909.
Warning: Impossible A1 allele assignment for variant rs3800829.
Warning: Impossible A1 allele assignment for variant rs7856222.
Warning: Impossible A1 allele assignment for variant rs3808902.
Warning: Impossible A1 allele assignment for variant rs17269854.
Warning: Impossible A1 allele assignment for variant rs534721.
Warning: Impossible A1 allele assignment for variant rs11139569.
Warning: Impossible A1 allele assignment for variant rs16908089.
Warning: Impossible A1 allele assignment for variant rs2245389.
Warning: Impossible A1 allele assignment for variant rs34312136.
Warning: Impossible A1 allele assignment for variant rs4078122.
Warning: Impossible A1 allele assignment for variant rs3207775.
Warning: Impossible A1 allele assignment for variant rs561415.
Warning: Impossible A1 allele assignment for variant rs11813861.
Warning: Impossible A1 allele assignment for variant rs1650166.
Warning: Impossible A1 allele assignment for variant rs11196005.
Warning: Impossible A1 allele assignment for variant rs12243523.
Warning: Impossible A1 allele assignment for variant rs7100377.
Warning: Impossible A1 allele assignment for variant rs12415539.
Warning: Impossible A1 allele assignment for variant rs2273748.
Warning: Impossible A1 allele assignment for variant rs5030779.
Warning: Impossible A1 allele assignment for variant rs516761.
Warning: Impossible A1 allele assignment for variant rs234872.
Warning: Impossible A1 allele assignment for variant rs7107290.
Warning: Impossible A1 allele assignment for variant rs4287314.
Warning: Impossible A1 allele assignment for variant rs10501259.
Warning: Impossible A1 allele assignment for variant rs4756057.
Warning: Impossible A1 allele assignment for variant rs1044796.
Warning: Impossible A1 allele assignment for variant rs17507049.
Warning: Impossible A1 allele assignment for variant rs661124.
Warning: Impossible A1 allele assignment for variant rs1894080.
Warning: Impossible A1 allele assignment for variant rs2324509.
Warning: Impossible A1 allele assignment for variant rs4936260.
Warning: Impossible A1 allele assignment for variant rs11524965.
Warning: Impossible A1 allele assignment for variant rs2159347.
Warning: Impossible A1 allele assignment for variant rs3782598.
Warning: Impossible A1 allele assignment for variant rs1451772.
Warning: Impossible A1 allele assignment for variant rs10505915.
Warning: Impossible A1 allele assignment for variant rs3782514.
Warning: Impossible A1 allele assignment for variant rs10772153.
Warning: Impossible A1 allele assignment for variant rs2372379.
Warning: Impossible A1 allele assignment for variant rs803569.
Warning: Impossible A1 allele assignment for variant rs2279405.
Warning: Impossible A1 allele assignment for variant rs6633.
Warning: Impossible A1 allele assignment for variant rs7957839.
Warning: Impossible A1 allele assignment for variant rs1413155.
Warning: Impossible A1 allele assignment for variant rs9300901.
Warning: Impossible A1 allele assignment for variant rs9604511.
Warning: Impossible A1 allele assignment for variant rs7996853.
Warning: Impossible A1 allele assignment for variant rs7399982.
Warning: Impossible A1 allele assignment for variant rs9604529.
Warning: Impossible A1 allele assignment for variant rs11259844.
Warning: Impossible A1 allele assignment for variant rs11618091.
Warning: Impossible A1 allele assignment for variant rs6602901.
Warning: Impossible A1 allele assignment for variant rs9577914.
Warning: Impossible A1 allele assignment for variant rs9604566.
Warning: Impossible A1 allele assignment for variant rs7323426.
Warning: Impossible A1 allele assignment for variant rs6602905.
Warning: Impossible A1 allele assignment for variant rs7323932.
Warning: Impossible A1 allele assignment for variant rs7996145.
Warning: Impossible A1 allele assignment for variant rs7332546.
Warning: Impossible A1 allele assignment for variant rs9550238.
Warning: Impossible A1 allele assignment for variant rs7400267.
Warning: Impossible A1 allele assignment for variant rs6602894.
Warning: Impossible A1 allele assignment for variant rs7399469.
Warning: Impossible A1 allele assignment for variant rs6602895.
Warning: Impossible A1 allele assignment for variant rs6422414.
Warning: Impossible A1 allele assignment for variant rs7335819.
Warning: Impossible A1 allele assignment for variant rs13379029.
Warning: Impossible A1 allele assignment for variant rs4147557.
Warning: Impossible A1 allele assignment for variant rs10483432.
Warning: Impossible A1 allele assignment for variant rs11627089.
Warning: Impossible A1 allele assignment for variant rs17597295.
Warning: Impossible A1 allele assignment for variant rs12793.
Warning: Impossible A1 allele assignment for variant rs1744296.
Warning: Impossible A1 allele assignment for variant rs4983517.
Warning: Impossible A1 allele assignment for variant rs8034978.
Warning: Impossible A1 allele assignment for variant rs3809581.
Warning: Impossible A1 allele assignment for variant rs10519208.
Warning: Impossible A1 allele assignment for variant rs3087567.
Warning: Impossible A1 allele assignment for variant rs2107234.
Warning: Impossible A1 allele assignment for variant rs252304.
Warning: Impossible A1 allele assignment for variant rs8061401.
Warning: Impossible A1 allele assignment for variant rs13337562.
Warning: Impossible A1 allele assignment for variant rs28437095.
Warning: Impossible A1 allele assignment for variant rs9928892.
Warning: Impossible A1 allele assignment for variant rs1048149.
Warning: Impossible A1 allele assignment for variant rs2306270.
Warning: Impossible A1 allele assignment for variant rs764688.
Warning: Impossible A1 allele assignment for variant rs3764420.
Warning: Impossible A1 allele assignment for variant rs7210126.
Warning: Impossible A1 allele assignment for variant rs17673149.
Warning: Impossible A1 allele assignment for variant rs11868321.
Warning: Impossible A1 allele assignment for variant rs1042678.
Warning: Impossible A1 allele assignment for variant rs3744155.
Warning: Impossible A1 allele assignment for variant rs16942082.
Warning: Impossible A1 allele assignment for variant rs9973085.
Warning: Impossible A1 allele assignment for variant rs11080748.
Warning: Impossible A1 allele assignment for variant rs355311.
Warning: Impossible A1 allele assignment for variant rs3826608.
Warning: Impossible A1 allele assignment for variant rs10502668.
Warning: Impossible A1 allele assignment for variant rs4919838.
Warning: Impossible A1 allele assignment for variant rs4523.
Warning: Impossible A1 allele assignment for variant rs11085099.
Warning: Impossible A1 allele assignment for variant rs3093088.
Warning: Impossible A1 allele assignment for variant rs14129.
Warning: Impossible A1 allele assignment for variant rs8100232.
Warning: Impossible A1 allele assignment for variant rs8109833.
Warning: Impossible A1 allele assignment for variant rs430989.
Warning: Impossible A1 allele assignment for variant rs3760667.
Warning: Impossible A1 allele assignment for variant rs3187346.
Warning: Impossible A1 allele assignment for variant rs2286750.
Warning: Impossible A1 allele assignment for variant rs6110212.
Warning: Impossible A1 allele assignment for variant rs3746600.
Warning: Impossible A1 allele assignment for variant rs3761210.
Warning: Impossible A1 allele assignment for variant rs17001274.
Warning: Impossible A1 allele assignment for variant rs17114359.
Warning: Impossible A1 allele assignment for variant rs3788014.
Warning: Impossible A1 allele assignment for variant rs35829851.
Warning: Impossible A1 allele assignment for variant rs2268780.
Warning: Impossible A1 allele assignment for variant rs4820280.
Warning: Impossible A1 allele assignment for variant rs9611591.
Warning: Impossible A1 allele assignment for variant rs34420568.
Warning: Impossible A1 allele assignment for variant rs9614750.
Warning: Impossible A1 allele assignment for variant rs34315830.
Warning: Impossible A1 allele assignment for variant rs5767487.
Warning: Impossible A1 allele assignment for variant rs35812349.
--a1-allele: 376380 assignments made.
376380 variants and 109 people pass filters and QC.
Among remaining phenotypes, 54 are cases and 55 are controls.
--make-bed to Results/GWAS4/HapMap-adj.bed + Results/GWAS4/HapMap-adj.bim +
Results/GWAS4/HapMap-adj.fam ... done.
2. To resolve strand issues, we flip SNPs found in both datasets with complementary alleles (i.e. they were reported in opposite strands). We generate SNP lists (ID and alleles) for both datasets, identify unique SNPs, and visualize differences in allele reporting. If a SNP is unique but reports alleles differently, it will appear twice. Below are examples of SNPs with strand issues from the 1000GP and HapMap data:
#print SNP and A1 A2 alleles on files for the two datasetsawk'{print$2,$5,$6}' Results/GWAS4/1kG_MDS5.bim > Results/GWAS4/1kGMDS5_tmpawk'{print$2,$5,$6}' Results/GWAS4/HapMap-adj.bim > Results/GWAS4/HapMap-adj_tmp#sort by SNP name to see SNPs with complementary allelessort Results/GWAS4/1kGMDS5_tmp Results/GWAS4/HapMap-adj_tmp |uniq-u> Results/GWAS4/all_differences.txt
head-6 Results/GWAS4/all_differences.txt
rs10006274 C T
rs10006274 G A
rs1008660 A G
rs1008660 T C
rs10088098 C T
rs10088098 G A
How many of these differences are there? We can count 604 lines, so 302 SNPs
wc-l Results/GWAS4/all_differences.txt
604 Results/GWAS4/all_differences.txt
Some of these differences might be might be due to strand issues.
Let’s look at this variant rs10006274. Will it be flipped in the HapMap dataset?
The answer is yes! If we look at the reference allele in 1kg_ref-list.txt, it shows C. This means the SNP is on the forward strand in 1000GP (C/T) and on the reverse strand in HapMap (G/A).
grep rs10006274 Results/GWAS4/1kg_ref-list.txt
rs10006274 C
grep rs10006274 Results/GWAS4/all_differences.txt
rs10006274 C T
rs10006274 G A
Stop - Read - Solve
Look at these other SNPs: rs9614750 and rs10088098.
Which ones will have to be flipped?
Is it always the same dataset that must be flipped?
# Write your code here
Solution
We will first print out the SNPs from the reference file to know which line corresponds to each dataset (since we know we used the 1000 Genomes Project as the reference). If there are strand issues, the SNP will need to be flipped in the dataset that wasn’t used as the reference.
grep rs9614750 Results/GWAS4/1kg_ref-list.txt
rs9614750 A
grep rs9614750 Results/GWAS4/all_differences.txt
rs9614750 A G
rs9614750 C G
For rs9614750, the genotype is reported as A/G in the 1000GP data, while in HapMap, it is C/G. This discrepancy between the two datasets means that the SNP will need to be removed later.
grep rs10088098 Results/GWAS4/1kg_ref-list.txt
rs10088098 C
grep rs10088098 Results/GWAS4/all_differences.txt
rs10088098 C T
rs10088098 G A
For rs10088098, the genotype is reported as C/T in the 1000GP data, while in HapMap, it is G/A. This means that PLINK will flip the allele, as they are complementary.
Now we take only the SNP names and give them to PLINK (option --flip), together with the reference genome (option --reference-allele):
## Flip SNPs for resolving strand issues.# Print SNP-identifier and remove duplicates.awk'{print$1}' Results/GWAS4/all_differences.txt |sort-u> Results/GWAS4/flip_list.txt
wc-l Results/GWAS4/flip_list.txt
302 Results/GWAS4/flip_list.txt
These are the SNP ID of non-corresponding SNPs (N=302) between the two files.
rs10006274 was flipped and has the same strand orientation in both datasets (same alleles in A1 and A2).
What happened to this one? PLINK attempts to resolve the mismatch by flipping the alleles but throws an error because the complementary alleles do not match the reference!
grep rs9614750 Results/GWAS4/corrected_hapmap.bim
22 rs9614750 0 44436371 G C
grep rs9614750 Results/GWAS4/HapMap-adj.bim
22 rs9614750 0 44436371 C G
Note
You don’t need to flip the 1000GP data because the reference allele (A1) in the 1000GP data already matches the strand orientation used in the HapMap data.
3. After flipping SNPs, some differ in their alleles when comparing datasets to each other (e.g. SNP rs9614750) and such SNPs must be removed.
We extract the SNPs from the corrected HapMap data and search for unique SNP (ID, A1, and A2), comparing them with those from the 1000GP data.
Perform MDS on HapMap-CEU data anchored by 1000 Genomes data.
MDS is typically performed on independent SNPs (pruned SNPs). We have previously identified such SNPs in this course, so we will extract only these SNPs for the analysis.
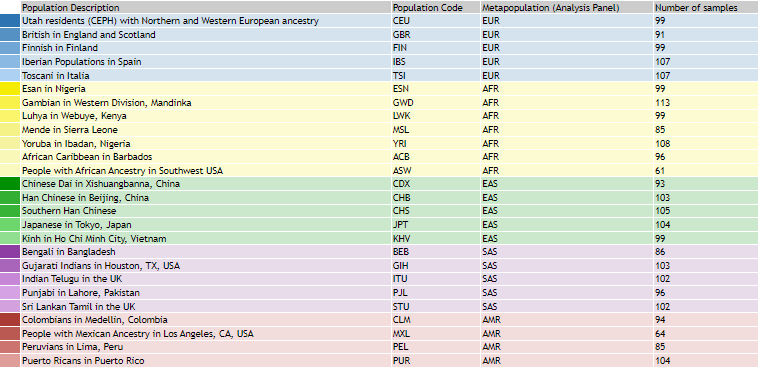
For visualization purposes, we downloaded the 1000 Genomes Project (1000GP) panel, which includes individual names and their corresponding population information.
To determine the population origins of HapMap individuals, we created a new file that combines the 1000GP panel information with the HapMap data, labeling the population HapMap entries as ‘OWN’.
The 1000 Genomes Project (1000GP) categorizes individuals into major continental groups—such as Europeans (EUR), Africans (AFR), Americans (AMR), East Asians (EAS), and South Asians (SAS)—each comprising various subpopulations. We will use this population structure information to visualize and determine the clusters our samples belong to.
Switch to the R kernel.
Let’s visualize population stratification using the multidimensional scaling (MDS) results.
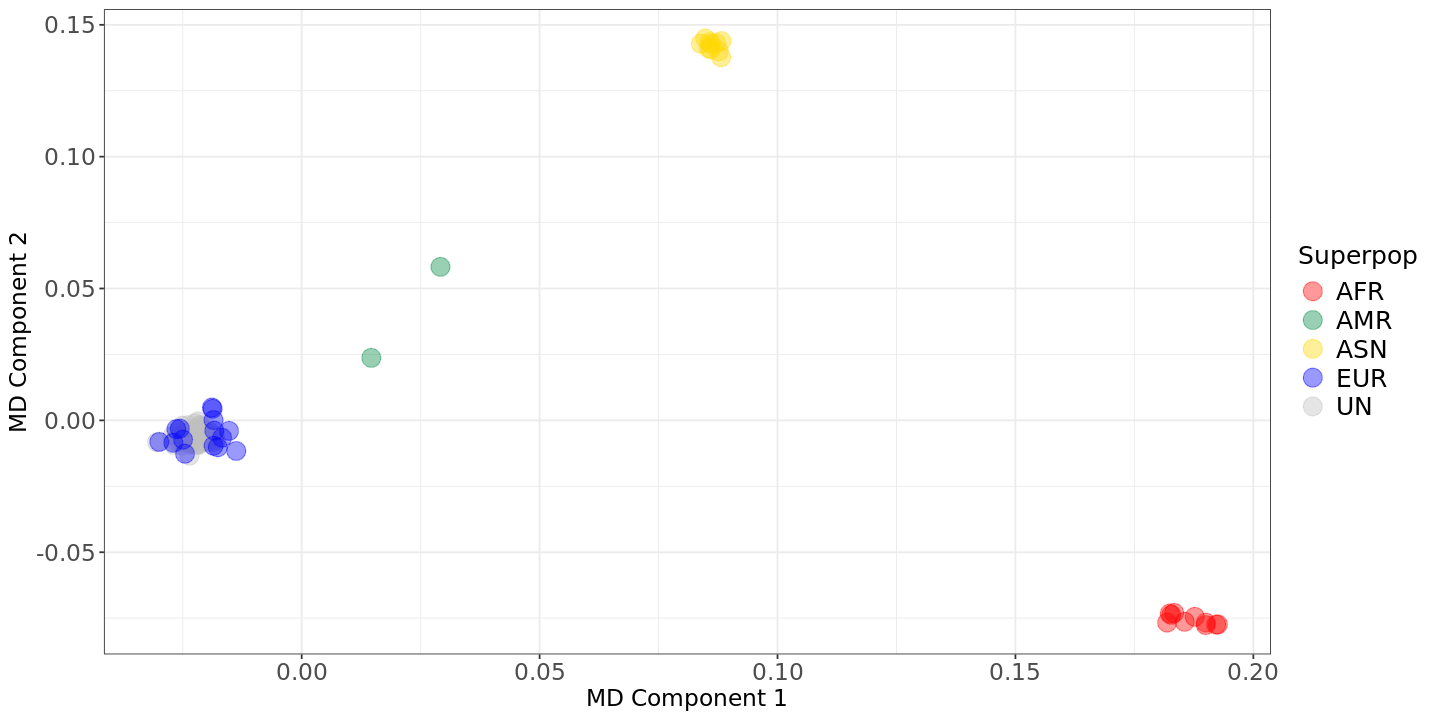
options(repr.plot.width = 12, repr.plot.height = 6)suppressMessages(suppressWarnings(library(ggplot2)))# Read data into R data<- read.table(file="Results/GWAS4/MDS_merge.mds",header=TRUE)pop<- read.table(file="Results/GWAS4/popfile.txt",header=TRUE)datafile<- merge(data,pop,by=c("FID","IID"))# Metapopulation information for the population in the 1000GP datasetsuperpop<- c("JPT" = "ASN","ASW" = "AFR","CEU" = "EUR","CHB" = "ASN","CHD" = "ASN","YRI" = "AFR","LWK" = "AFR","TSI" = "EUR","MXL" = "AMR","GBR" = "EUR","FIN" = "EUR","CHS" = "ASN","PUR" = "AMR","OWN" = "UN")# add metapopulation info to the table datafile$SUPERPOP<- superpop[datafile$POP]# Plotting scatter.mds<- ggplot(datafile, aes(x=C1, y=C2, color=SUPERPOP))+geom_point(size=5, alpha=.4)+scale_color_manual(values=c("AFR" = "red", "AMR" = "springgreen4", "ASN" = "gold", "EUR" = "blue", "UN" = "grey"))+xlab("MD Component 1")+ylab("MD Component 2")+labs(color="Superpop")+theme_bw()+theme(axis.title = element_text(size = 14), legend.text = element_text(size = 15),axis.text = element_text(size = 14), legend.title=element_text(size=15))show(scatter.mds)
The HapMap data clusters closely with European populations such as CEU, TSI, IBS, GBR, and FIN, confirming its European composition. Additionally, the absence of distant points indicates no outliers in the HapMap dataset (grey datapoints cluster together).
Exclude ethnic outliers
To control for population structure, individuals that deviate substantially from their genetically inferred ancestry cluster are often excluded. A common approach is to calculate genetic principal components (PCs) and compute each individual’s Mahalanobis or Euclidean distance from the center of their assigned cluster; individuals beyond a predefined threshold (e.g., >4 standard deviations) are considered population outliers and removed from downstream analysis.
Now, imagine we want to consider only European individuals for a specific analysis where we want only such a homogeneous population. Then the rest of the individuals would be considered outliers.
Stop - Read - Solve
Based on the PCA plot, visually inspect the distribution of individuals along the top principal components and determine approximate threshold values beyond which samples should be considered outliers and excluded. What cutoff values would you apply in this context, and why?
Solution
Based on the position of the target population’s cluster in the MDS plot, we will exclude individuals that fall outside the European cluster in the PCA.
Switch to the Bash kernel. Let’s see how the MDS table is structured:
The first three columns denote samples, while C1, C2, … are MDS components. We will select the individuals we want to keep based on coordinates C1, C2, so the area to be chosen has to be C1<0 and C2<0.05. Thus we take the values in columns 4 and 5 of the file , and extract the individuals using PLINK with the option --keep.
#print samples to keep in a fileawk'{ if ($4 <0 && $5<0.05) print $1,$2 }' Results/GWAS4/MDS_merge.mds > Results/GWAS4/EUR_MDS_merge#use the keep --optionplink--bfile Results/GWAS4/MDS_merge \--keep Results/GWAS4/EUR_MDS_merge \--make-bed\--out Results/GWAS4/EUR_MDS_merge
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/EUR_MDS_merge.log.
Options in effect:
--bfile Results/GWAS4/MDS_merge
--keep Results/GWAS4/EUR_MDS_merge
--make-bed
--out Results/GWAS4/EUR_MDS_merge
385567 MB RAM detected; reserving 192783 MB for main workspace.
376380 variants loaded from .bim file.
146 people (55 males, 54 females, 37 ambiguous) loaded from .fam.
Ambiguous sex IDs written to Results/GWAS4/EUR_MDS_merge.nosex .
109 phenotype values loaded from .fam.
--keep: 124 people remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 124 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate in remaining samples is 0.99826.
376380 variants and 124 people pass filters and QC.
Among remaining phenotypes, 54 are cases and 55 are controls. (15 phenotypes
are missing.)
--make-bed to Results/GWAS4/EUR_MDS_merge.bed + Results/GWAS4/EUR_MDS_merge.bim
+ Results/GWAS4/EUR_MDS_merge.fam ... done.
How many people are we left with? see the output message above or find the log file
grep"remaining" Results/GWAS4/EUR_MDS_merge.log
--keep: 124 people remaining.
Total genotyping rate in remaining samples is 0.99826.
Among remaining phenotypes, 54 are cases and 55 are controls. (15 phenotypes
Creating covariates for GWAS analysis
The 10 MDS dimensions will be used as covariates in the association analysis in the next tutorial to correct for population stratification. The covariate file is created by removing column 3 (SOL, optional metadata) from the MDS output file.
Stop - Read - Solve
Why are we computing the covariates again?
Solution
The data has been filtered, and the remaining samples will have a very tight MDS structure, since they were projected in relationship to other very different samples. Now we might want to capture some substructure in the CEU population instead.
This time we will not use MDS projection, but the PCA implemented in PLINK2. In our dataset this will not change much (actually it might be a bit slower, because it also recalculates the relatedness matrix and normalizes it), but for large datasets PCA is faster, and you can also use an approximated PCA which is even faster on many thousands of samples (all details here in the manual). The default is to calculate ten components, but you can for example choose more as we fo here. biallelic-var-wts ensures PLINK2 does not apply its own filtering to our data.
PLINK v2.00a5.12LM 64-bit Intel (25 Jun 2024) www.cog-genomics.org/plink/2.0/
(C) 2005-2024 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to Results/GWAS4/EUR_MDS_merge.log.
Options in effect:
--bfile Results/GWAS4/EUR_MDS_merge
--out Results/GWAS4/EUR_MDS_merge
--pca 20 biallelic-var-wts
Start time: Tue Apr 1 10:22:19 2025
385567 MiB RAM detected, ~357174 available; reserving 192783 MiB for main
workspace.
Using up to 64 threads (change this with --threads).
124 samples (54 females, 55 males, 15 ambiguous; 124 founders) loaded from
Results/GWAS4/EUR_MDS_merge.fam.
376380 variants loaded from Results/GWAS4/EUR_MDS_merge.bim.
1 binary phenotype loaded (54 cases, 55 controls).
Calculating allele frequencies... done.
Constructing GRM: done.
Correcting for missingness... done.
Extracting eigenvalues and eigenvectors... done.
--pca: Variant weights written to Results/GWAS4/EUR_MDS_merge.eigenvec.var .
--pca: Eigenvectors written to Results/GWAS4/EUR_MDS_merge.eigenvec , and
eigenvalues written to Results/GWAS4/EUR_MDS_merge.eigenval .
End time: Tue Apr 1 10:22:30 2025
The output file looks like this, where the PCA components start from the third columns
Switch to the R kernel. Let’s print the PCA again.
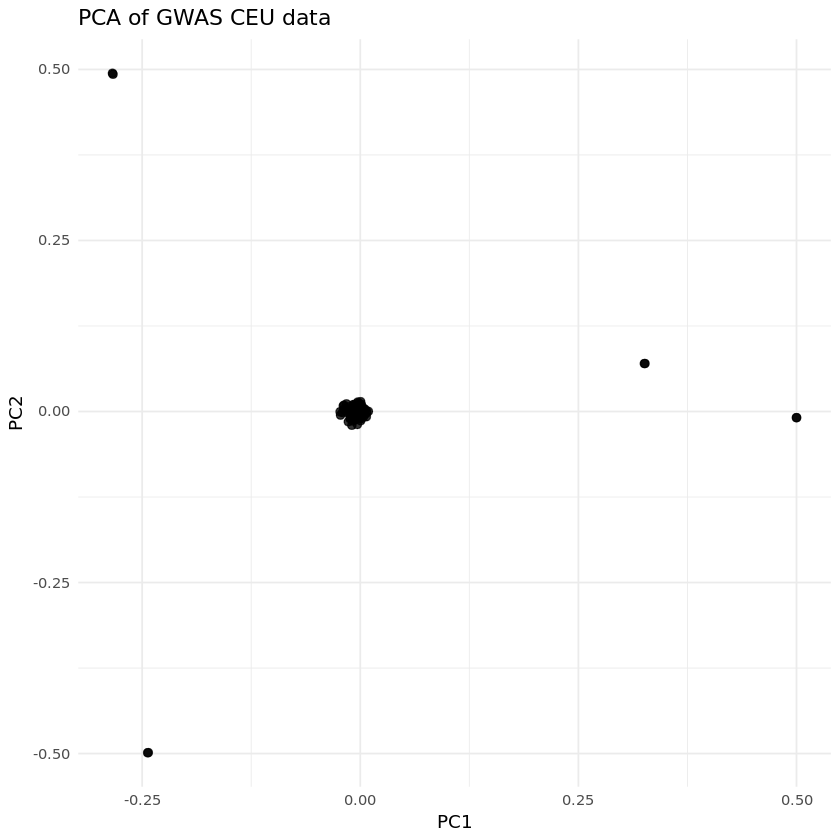
library(ggplot2)library(data.table)# For fast data reading# Load the PCA eigenvector filepca_data<- fread("Results/GWAS4/EUR_MDS_merge.eigenvec", header = TRUE)# Preview the datahead(pca_data)dim(pca_data)
A data.table: 6 × 22
#FID
IID
PC1
PC2
PC3
PC4
PC5
PC6
PC7
PC8
⋯
PC11
PC12
PC13
PC14
PC15
PC16
PC17
PC18
PC19
PC20
<chr>
<chr>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
⋯
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
<dbl>
1328
NA06984
-0.00859205
-0.000713344
1.94880e-03
-0.0194148
0.00557532
-0.00846987
-0.11407800
-0.01715050
⋯
0.10400200
-0.05559300
0.07365730
-0.01352440
-0.03648780
0.00538918
-0.10914400
-0.05936100
-0.004409370
-0.04319370
1328
NA06989
-0.01366580
-0.014994200
3.01289e-03
-0.0254053
0.27208800
0.00254102
0.14493300
0.11984600
⋯
-0.06302050
0.03569980
-0.07596030
-0.03705540
-0.04184730
0.05273180
-0.01213660
0.04795970
-0.036786700
-0.03056990
1330
NA12340
-0.00616811
-0.004852810
-4.46531e-04
-0.0206360
-0.03074260
0.07586640
0.01315840
0.02814490
⋯
-0.00743298
-0.00302273
-0.06983250
0.08204050
0.09200260
0.11563000
-0.02472290
0.09715490
0.246957000
-0.02117160
1330
NA12341
0.00463878
0.001408340
1.10760e-02
-0.0232280
-0.08110370
0.01636190
-0.00248169
0.04314810
⋯
0.07873880
0.05197300
0.01933570
-0.01668130
0.03402380
-0.02396450
0.14352700
-0.03609260
0.024535600
0.09993830
1330
NA12343
0.00317760
-0.004503100
-8.72381e-05
-0.0163116
-0.02494240
-0.01609400
-0.00862269
0.04075270
⋯
0.09798020
0.04510380
-0.01979330
-0.06638900
0.02757650
-0.04051890
-0.03975800
-0.05994600
-0.008285970
-0.13435200
1334
NA12144
-0.24370100
-0.498832000
-3.15505e-02
0.4248900
-0.00277974
-0.00238691
-0.01056630
-0.00688372
⋯
-0.00439512
-0.00402327
-0.00216514
-0.00551774
0.00505487
-0.00129990
-0.00459691
0.00525804
-0.000930452
0.00247628
124
22
We can see some substructure, where most individuals are tightly clustered together.
You have now successfully checked your data for relatedness population stratification. You filtered out the individuals with high relatedness and produced a summary of the population structure using the MDS projection. You will use the MDS coordinates as a proxy for the population structure you want your association testing to be corrected for.
In the next notebook on Association Testing, you will need the following files from the folder Results/GWAS4/:
EUR_MDS_merge PLINK file set
covar_pca.txt
which are the HapMap data and the MDS covariates highlighting the population stratification inside the european cluster. Those are already available once you have been running this notebook.
Below is a cheat sheet of our new methods of QC.
Step
Command
Function
Thresholds and explanation
7: Population Stratification
--cluster --mds-plot k (PLINK)
Produces a k‐dimensional representation of any substructure in the data, based on IBS.
K is the number of dimensions, which needs to be defined (typically 10). This is an important step of the QC that consists of multiple proceedings but for reasons of completeness, we briefly refer to this step in the table. This step will be described in more detail in the section “Controlling for population stratification.”
-
--pca k biallelic-var-wts (PLINK2)
produces a PCA projection with k principal components, based on the normalized genetic relationship matrix. biallelic-var-wts ensures that PLINK2’s own filtering is not applied.
-
Bibliography
Abdellaoui, Abdel, Jouke-Jan Hottenga, Peter de Knijff, Michel G Nivard, Xiangjun Xiao, Paul Scheet, Andrew Brooks, et al. 2013. “Population Structure, Migration, and Diversifying Selection in the Netherlands.”European Journal of Human Genetics 21 (11): 1277–85.
Cortellari, Matteo, Mario Barbato, Andrea Talenti, Arianna Bionda, Antonello Carta, Roberta Ciampolini, Elena Ciani, et al. 2021. “The Climatic and Genetic Heritage of Italian Goat Breeds with Genomic SNP Data.”Scientific Reports 11 (1): 10986. https://doi.org/10.1038/s41598-021-89900-2.
Price, Alkes L., Nick J. Patterson, Robert M. Plenge, Michael E. Weinblatt, Nancy A. Shadick, and David Reich. 2006. “Principal Components Analysis Corrects for Stratification in Genome-Wide Association Studies.”Nature Genetics 38 (8): 904–9. https://doi.org/10.1038/ng1847.
Price, Alkes L, Noah A Zaitlen, David Reich, and Nick Patterson. 2010. “New Approaches to Population Stratification in Genome-Wide Association Studies.”Nature Reviews Genetics 11 (7): 459–63.
Rietveld, Cornelius A, Sarah E Medland, Jaime Derringer, Jian Yang, Tõnu Esko, Nicolas W Martin, Harm-Jan Westra, et al. 2013. “GWAS of 126,559 Individuals Identifies Genetic Variants Associated with Educational Attainment.”Science 340 (6139): 1467–71.
Somers, Metten, Loes M. Olde Loohuis, Maartje F. Aukes, Bogdan Pasaniuc, Kees C. L. De Visser, René S. Kahn, Iris E. Sommer, and Roel A. Ophoff. 2017. “A GeneticPopulationIsolate in TheNetherlandsShowingExtensiveHaplotypeSharing and LongRegions of Homozygosity.”Genes 8 (5): 133. https://doi.org/10.3390/genes8050133.
 Choose the Bash kernel
Choose the Bash kernel