# DO NOT RUN
# Create dds object
dds <- DESeqDataSetFromTximport(txi,
colData = meta %>% column_to_rownames("sample"),
design = ~ condition)
# Filter genes with 0 counts
keep <- rowSums(counts(dds)) > 0
dds <- dds[keep,]Gene-level differential expression analysis with DESeq2
Approximate time: 15 minutes
Learning Objectives
- Explain the different steps involved in running
DESeq() - Examine size factors and understand the source of differences
- Inspect gene-level dispersion estimates
- Recognize the importance of dispersion during differential expression analysis
DESeq2 differential gene expression analysis workflow
Previously, we created the DESeq2 object using the appropriate design formula.
Then, to run the entire differential expression analysis workflow, we use a single call to the function DESeq().
## Run analysis
dds <- DESeq(dds)And with that we completed the entire workflow for the differential gene expression analysis with DESeq2! The DESeq() function performs a default analysis through the following steps:
- Estimation of size factors:
estimateSizeFactors() - Estimation of dispersion:
estimateDispersions() - Negative Binomial GLM fitting and Wald statistics:
nbinomWaldTest()
Step 1: Estimate size factors
The first step in the differential expression analysis is to estimate the size factors, which is exactly what we already did to normalize the raw counts.
Let’s take a quick look at size factor values we have for each sample:
## Check the size factors
sizeFactors(dds) control_3 control_2 control_1 vampirium_3 vampirium_2 vampirium_1
0.7501070 0.9580056 1.1134126 0.6565921 1.1418067 1.2184977
garlicum_3 garlicum_2
0.9327493 1.5527069 These numbers should be identical to those we generated initially when we had run the function estimateSizeFactors(dds). Take a look at the total number of reads for each sample:
## Total number of raw counts per sample
colSums(counts(dds)) control_3 control_2 control_1 vampirium_3 vampirium_2 vampirium_1
20257917 26156069 30817090 17054657 29573402 31589516
garlicum_3 garlicum_2
26428310 44740987 How do the numbers correlate with the size factor?
We see that the larger size factors correspond to the samples with higher sequencing depth, which makes sense, because to generate our normalized counts we need to divide the counts by the size factors. This accounts for the differences in sequencing depth between samples.
Now take a look at the total depth after normalization using:
## Total number of normalized counts per sample
colSums(counts(dds, normalized=T)) control_3 control_2 control_1 vampirium_3 vampirium_2 vampirium_1
27006701 27302628 27678052 25974507 25900533 25924969
garlicum_3 garlicum_2
28333777 28814831 How do the values across samples compare with the total counts taken for each sample?
You might have expected the counts to be the exact same across the samples after normalization. However, DESeq2 also accounts for RNA composition during the normalization procedure. By using the median ratio value for the size factor, DESeq2 should not be biased to a large number of counts sucked up by a few DE genes; however, this may lead to the size factors being quite different than what would be anticipated just based on sequencing depth.
Step 2: Estimate gene-wise dispersion
Let’s take a look at the dispersion estimates for our Vampirium data. First, we will use the function estimateDispersions().
dds <- estimateDispersions(dds)We can check the values using the dispersion() function and plotting it with the plotDispEsts() function:
head(dispersions(dds))[1] 0.102415719 0.005312987 0.005241663 9.982833068 0.954109932 0.002146811## Plot dispersion estimates
plotDispEsts(dds)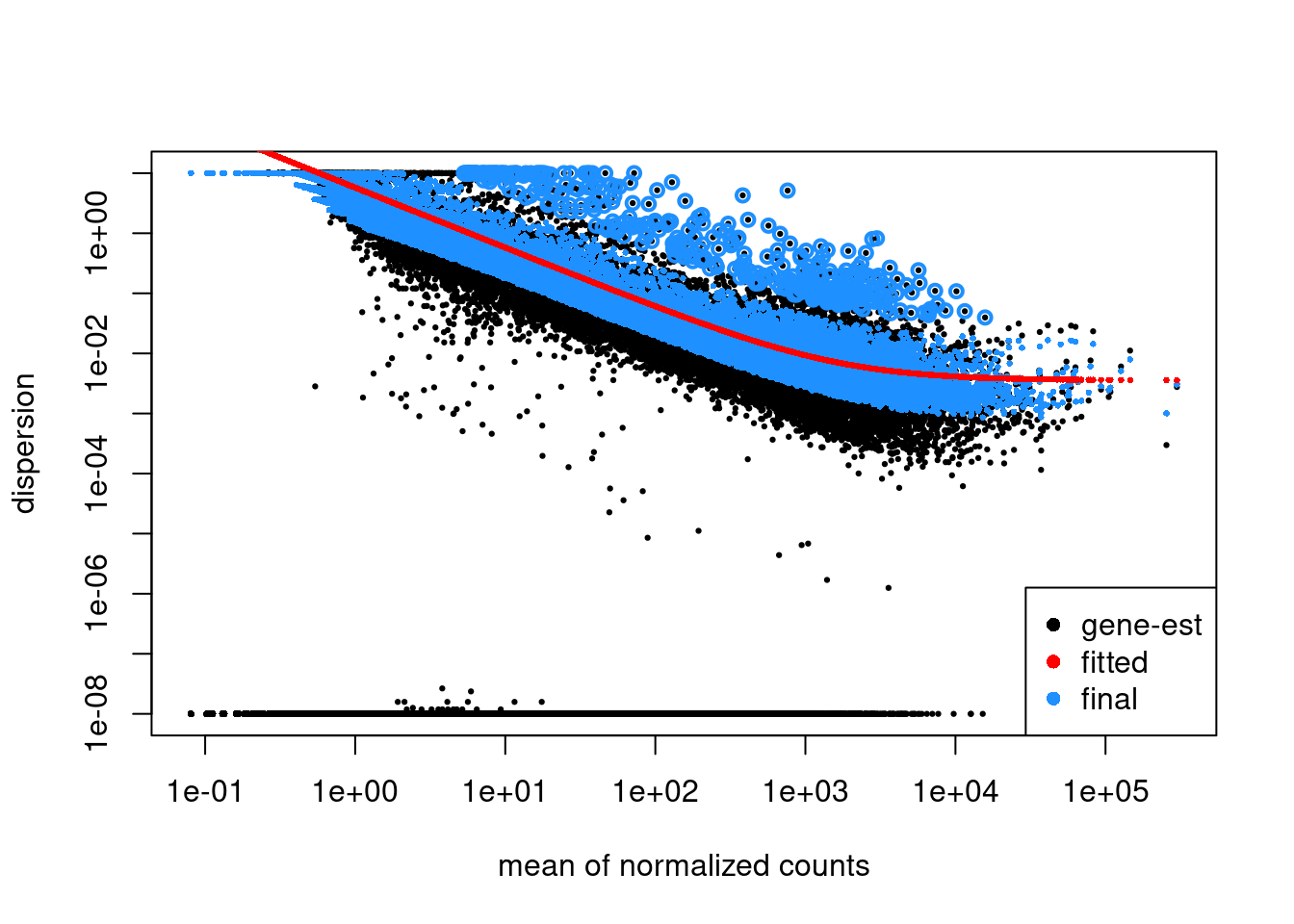
We can see that our estimated dispersions look quite good!
Exercise 1
Given the dispersion plot below, would you have any concerns regarding the fit of your data to the model?
- If not, what aspects of the plot makes you feel confident about your data?
- If so, what are your concerns? What would you do to address them?