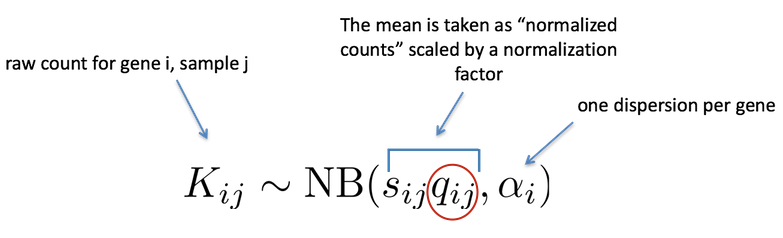
Hypothesis testing with DESeq2
Approximate time: 40 minutes
Learning Objectives
- Discuss the steps required to generate a results table for pairwise comparisons (Wald test)
- Recognize the importance of multiple test correction
- Identify different methods for multiple test correction
- Summarize the different levels of gene filtering
- Evaluate the number of differentially expressed genes produced for each comparison
- Construct R objects containing significant genes from each comparison
DESeq2: Model fitting and Hypothesis testing
The final step in the DESeq2 workflow is taking the counts for each gene and fitting it to the model and testing for differential expression.
Generalized Linear Model
As described earlier, the count data generated by RNA-seq exhibits overdispersion (variance > mean) and the statistical distribution used to model the counts needs to account for this. As such, DESeq2 uses a negative binomial distribution to model the RNA-seq counts using the equation below:
The two parameters required are the size factor, and the dispersion estimate. Next, a generalized linear model (GLM) of the NB family is used to fit the data. Modeling is a mathematically formalized way to approximate how the data behaves given a set of parameters.
After the model is fit, coefficients are estimated for each sample group along with their standard error. The coefficients are the estimates for the log2 fold changes, and will be used as input for hypothesis testing.
Hypothesis testing
The first step in hypothesis testing is to set up a null hypothesis for each gene. In our case, the null hypothesis is that there is no differential expression across the two sample groups (LFC == 0). Second, we use a statistical test to determine if based on the observed data, the null hypothesis can be rejected. In DESeq2, the Wald test is the default used for hypothesis testing when comparing two groups.
The GLM and the statistical testing is done using the function nbinomWaldTest()
dds <- nbinomWaldTest(dds)The model fit and Wald test were already run previously as part of the DESeq() function:
## DO NOT RUN THIS CODE
## Create DESeq2Dataset object
dds <- DESeqDataSetFromTximport(txi,
colData = meta %>% column_to_rownames("sample"),
design = ~ condition)
## Run analysis
dds <- DESeq(dds)Exploring Results
By default DESeq2 uses the Wald test to identify genes that are differentially expressed between two sample groups. Given the factor(s) used in the design formula, and how many factor levels are present, we can extract results for a number of different comparisons.
Specifying contrasts
In our dataset, we have three sample groups so we can make three possible pairwise comparisons:
- Control vs. Vampirium
- Garlicum vs. Vampirium
- Garlicum vs. Control
We are really only interested in #1 and #2 from above. When we initially created our dds object we had provided ~ condition as our design formula, indicating that condition is our main factor of interest.
To indicate which two sample groups we are interested in comparing, we need to specify contrasts. The contrasts are used as input to the DESeq2 results() function to extract the desired results.
Contrasts can be specified in three different ways:
- One of the results from
resultsNames(dds)and thenameargument. This one is the simplest but it can also be very restricted:
resultsNames(dds) # to see what names to useresults(dds, name = resultsNames(dds)[2]) # here we specify the second comparison from the list.- Contrasts can be supplied as a character vector with exactly three elements:
The first element design_var, is one of the variables you provided to the design formula on the DESeqDataSetFromTximport() function, when you first created the dds object.
The second element, level_to_compare, is your group of interest of the chosen variable. In our case it would be either Control samples or Garlicum samples. The group must match the name in your metadata.
The last element, base_level, is, ofc, the base level for the comparison. This would be our vampirium samples. Again, the name must match the name in your metadata!
# DO NOT RUN!
contrast <- c("design_var", "level_to_compare", "base_level")
results(dds, contrast = contrast)- Contrasts can be given as a list of 2 character vectors: the names of the fold changes for the level of interest, and the names of the fold changes for the base level. These names should match identically to the elements of
resultsNames(object). This method can be useful for combining interaction terms and main effects.
# DO NOT RUN!
resultsNames(dds) # to see what names to use
contrast <- list(resultsNames(dds)[2], resultsNames(dds)[3])
results(dds, contrast = contrast)Alternatively, if you only had two factor levels you could do nothing:
results(dds)log2 fold change (MLE): condition garlicum vs vampirium
Wald test p-value: condition garlicum vs vampirium
DataFrame with 38872 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000005 26.086073 -0.646861 0.501159 -1.290729 0.1967975
ENSG00000000419 1614.076845 -0.149505 0.100881 -1.481997 0.1383411
ENSG00000000457 509.265870 0.194316 0.109504 1.774518 0.0759776
ENSG00000000938 0.403756 1.805083 4.476581 0.403228 0.6867805
ENSG00000000971 8.381364 -1.884550 1.452071 -1.297836 0.1943437
... ... ... ... ... ...
ENSG00000291310 0.380754 -2.6717039 4.492548 -0.594697 0.552046156
ENSG00000291312 20.825465 -1.6970593 1.002445 -1.692920 0.090470680
ENSG00000291315 30.487909 -0.0768952 0.470023 -0.163599 0.870046849
ENSG00000291316 73.844923 0.7412490 0.342495 2.164264 0.030444100
ENSG00000291317 299.454400 -0.7642255 0.186602 -4.095488 0.000042128
padj
<numeric>
ENSG00000000005 0.383132
ENSG00000000419 0.299549
ENSG00000000457 0.193966
ENSG00000000938 NA
ENSG00000000971 0.379530
... ...
ENSG00000291310 NA
ENSG00000291312 0.220112811
ENSG00000291315 0.937261738
ENSG00000291316 0.097098441
ENSG00000291317 0.000404955To start, we want to evaluate expression changes between the Control samples and the Vampirium samples. As such we will use the first method for specifying contrasts and create a character vector:
Exercise 1
Define contrasts for Control vs Vampirium samples using one of the two methods above.
Your code here
contrast_cont <- The results table
Now that we have our contrast created, we can use it as input to the results() function.
?resultsYou will see we have the option to provide a wide array of arguments and tweak things from the defaults as needed. For example:
## Extract results for Contorl vs Vampirium with a pvalue < 0.05
res_tableCont <- results(dds, contrast=contrast_cont, alpha = 0.05)The results table that is returned to us is a DESeqResults object, which is a simple subclass of DataFrame.
# Check what type of object is returned
class(res_tableCont)Now let’s take a look at what information is stored in the results:
# What is stored in results?
res_tableCont %>%
data.frame() %>%
head()We can use the mcols() function to extract information on what the values stored in each column represent:
# Get information on each column in results
data.frame(mcols(res_tableCont, use.names=T))baseMean: mean of normalized counts for all samples in your count matrixlog2FoldChange: log2 fold changelfcSE: standard error for the lfc calculationstat: Wald statisticpvalue: Wald test p-valuepadj: BH adjusted p-values
Gene-level filtering
Let’s take a closer look at our results table. As we scroll through it, you will notice that for selected genes there are NA values in the pvalue and padj columns. What does this mean?
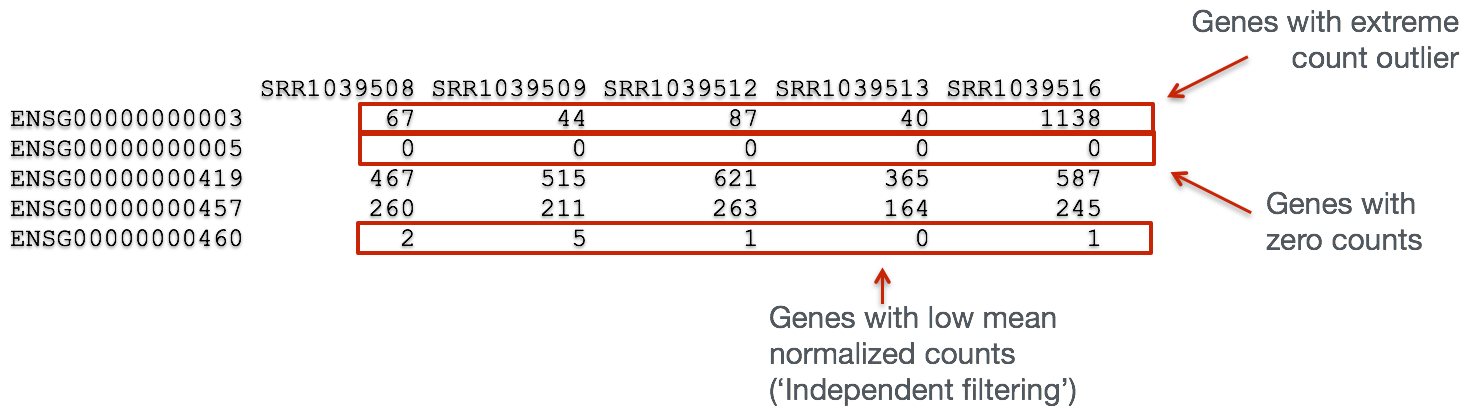
The missing values represent genes that have undergone filtering as part of the DESeq() function. Prior to differential expression analysis it is beneficial to omit genes that have little or no chance of being detected as differentially expressed. This will increase the power to detect differentially expressed genes. DESeq2 does not physically remove any genes from the original counts matrix, and so all genes will be present in your results table. The genes omitted by DESeq2 meet one of the three filtering criteria outlined below:
1. Genes with zero counts in all samples
If within a row, all samples have zero counts there is no expression information and therefore these genes are not tested. Since we have already filtered out these genes ourselves when we created our dds object.
# Show genes with zero expression
res_tableCont %>%
as_tibble(rownames = "gene") %>%
dplyr::filter(baseMean==0) %>%
head()If there would be any genes meeting this criteria, the baseMean column for these genes will be zero, and the log2 fold change estimates, p-value and adjusted p-value will all be set to NA.
2. Genes with an extreme count outlier
The DESeq() function calculates, for every gene and for every sample, a diagnostic test for outliers called Cook’s distance. If several samples are flagged for a certain gene, the gene is filtered out.
# Show genes that have an extreme outlier
res_tableCont %>%
as_tibble(rownames = "gene") %>%
dplyr::filter(is.na(pvalue) & is.na(padj) & baseMean > 0) %>%
head()It seems that we have some genes with outliers!
If a gene contains a sample with an extreme count outlier then the p-value and adjusted p-value will be set to NA.
3. Genes with a low mean normalized counts
DESeq2 defines a low mean threshold, that is empirically determined from your data, in which the fraction of significant genes can be increased by reducing the number of genes that are considered for multiple testing. This is based on the notion that genes with very low counts are not likely to see significant differences typically due to high dispersion.
# Show genes below the low mean threshold
res_tableCont %>%
as_tibble(rownames = "gene") %>%
dplyr::filter(!is.na(pvalue) & is.na(padj) & baseMean > 0) %>%
head()If a gene is filtered by independent filtering, then only the adjusted p-value will be set to NA.
NOTE: DESeq2 will perform the filtering outlined above by default; however other DE tools, such as EdgeR will not. Filtering is a necessary step, even if you are using limma-voom and/or edgeR’s quasi-likelihood methods. Be sure to follow pre-filtering steps when using other tools, as outlined in their user guides found on Bioconductor as they generally perform much better.
Fold changes
With large significant gene lists it can be hard to extract meaningful biological relevance. To help increase stringency, one can also add a fold change threshold. Keep in mind when setting that value that we are working with log2 fold changes, so a cutoff of log2FoldChange < 1 would translate to an actual fold change of 2.
An alternative approach to add the fold change threshold:
The
results()function has an option to add a fold change threshold using thelfcThresholdargument. This method is more statistically motivated, and is recommended when you want a more confident set of genes based on a certain fold-change. It actually performs a statistical test against the desired threshold, by performing a two-tailed test for log2 fold changes greater than the absolute value specified. The user can change the alternative hypothesis usingaltHypothesisand perform two one-tailed tests as well. This is a more conservative approach, so expect to retrieve a much smaller set of genes!
res_tableCont_LFC1 <- results(dds, contrast=contrast_cont, alpha = 0.05, lfcThreshold = 1)Summarizing results
To summarize the results table, a handy function in DESeq2 is summary().
## Summarize results
summary(res_tableCont, alpha = 0.05)In addition to the number of genes up- and down-regulated at the default threshold, the function also reports the number of genes that were tested (genes with non-zero total read count), and the number of genes not included in multiple test correction due to a low mean count.
Extracting significant differentially expressed genes
Let’s first create variables that contain our threshold criteria. We will only be using the adjusted p-values in our criteria:
### Set thresholds
padj.cutoff <- 0.05We can easily subset the results table to only include those that are significant using the dplyr::filter() function, but first we will convert the results table into a tibble:
# Create a tibble of results and add gene symbols to new object
res_tableCont_tb <- res_tableCont %>%
as_tibble(rownames = "gene") %>%
relocate(gene, .before = baseMean)
head(res_tableCont_tb)Now we can subset that table to only keep the significant genes using our pre-defined thresholds:
# Subset the tibble to keep only significant genes
sigCont <- res_tableCont_tb %>%
dplyr::filter(padj < padj.cutoff)# Take a quick look at this tibble
head(sigCont)Now that we have extracted the significant results, we are ready for visualization!
Exercise 2
Vampirium Differential Expression Analysis: Garlicum versus Vampirium
Now that we have results for the Control vs Vampirium results, do the same for the Garlicum vs. Control samples.
- Create a contrast vector called
contrast_gar. - Use contrast vector in the
results()to extract a results table and store that to a variable calledres_tableGar. - Using a p-adjusted threshold of 0.05 (
padj.cutoff < 0.05), subsetres_tableGarto report the number of genes that are up- and down-regulated in Garlicum compared to Vampirium. - How many genes are differentially expressed in the Garlicum vs Vampirium comparison? How does this compare to the Control vs Vampirium significant gene list (in terms of numbers)?