## Remove any NA values (reduces the data by quite a bit) and duplicates
res_entrez <- dplyr::filter(res_ids, entrez != "NA" & duplicated(entrez)==F)Functional Class Scoring
Approximate time: 30 minutes
Learning Objectives:
- Discuss functional class scoring
- Construct a GSEA analysis using GO and KEGG gene sets
- Examine results of a GSEA using pathview package
Gene set enrichment analysis using clusterProfiler and Pathview
Using the log2 fold changes obtained from the differential expression analysis for every gene, gene set enrichment analysis and pathway analysis can be performed using clusterProfiler and Pathview tools.
For a gene set or pathway analysis using clusterProfiler, coordinated differential expression over gene sets is tested instead of changes of individual genes. “Gene sets are pre-defined groups of genes, which are functionally related. Commonly used gene sets include those derived from KEGG pathways, Gene Ontology terms, MSigDB, Reactome, or gene groups that share some other functional annotations, etc. Consistent perturbations over such gene sets frequently suggest mechanistic changes”.
Preparation for GSEA
clusterProfiler offers several functions to perform GSEA using different genes sets, including but not limited to GO, KEGG, and MSigDb. We will use the KEGG gene sets, which identify genes using their Entrez IDs. Therefore, to perform the analysis, we will need to acquire the Entrez IDs. We will also need to remove the Entrez ID NA values and duplicates (due to gene ID conversion) prior to the analysis:
Finally, extract and name the fold changes:
## Extract the foldchanges
foldchanges <- res_entrez$log2FoldChange
## Name each fold change with the corresponding Entrez ID
names(foldchanges) <- res_entrez$entrezNext we need to order the fold changes in decreasing order. To do this we’ll use the sort() function, which takes a vector as input. This is in contrast to Tidyverse’s arrange(), which requires a data frame.
## Sort fold changes in decreasing order
foldchanges <- sort(foldchanges, decreasing = TRUE)
head(foldchanges) 1297 1501 100996732 107985200 3263 3934
4.182128 3.467430 3.280630 3.280630 3.001757 2.922119 Performing GSEA
To perform the GSEA using KEGG gene sets with clusterProfiler, we can use the gseKEGG() function:
## GSEA using gene sets from KEGG pathways
gseaKEGG <- gseKEGG(geneList = foldchanges, # ordered named vector of fold changes (Entrez IDs are the associated names)
organism = "hsa", # supported organisms listed below
pvalueCutoff = 0.05, # padj cutoff value
verbose = FALSE)
## Extract the GSEA results
gseaKEGG_results <- gseaKEGG@result
head(gseaKEGG_results)NOTE: The organisms with KEGG pathway information are listed here.
How many pathways are enriched?
View the enriched pathways:
## Write GSEA results to file
write.csv(gseaKEGG_results, "../Results/gsea_Cont-Vamp_kegg.csv", quote=F)NOTE: We will all get different results for the GSEA because the permutations performed use random reordering. If we would like to use the same permutations every time we run a function (i.e. we would like the same results every time we run the function), then we could use the
set.seed(123456)function prior to running. The input toset.seed()could be any number, but if you would want the same results, then you would need to use the same number as input.
Explore the GSEA plot of enrichment of one of the pathways in the ranked list:
## Plot the GSEA plot for a single enriched pathway:
gseaplot(gseaKEGG, geneSetID = gseaKEGG_results$ID[1], title = gseaKEGG_results$Description[1])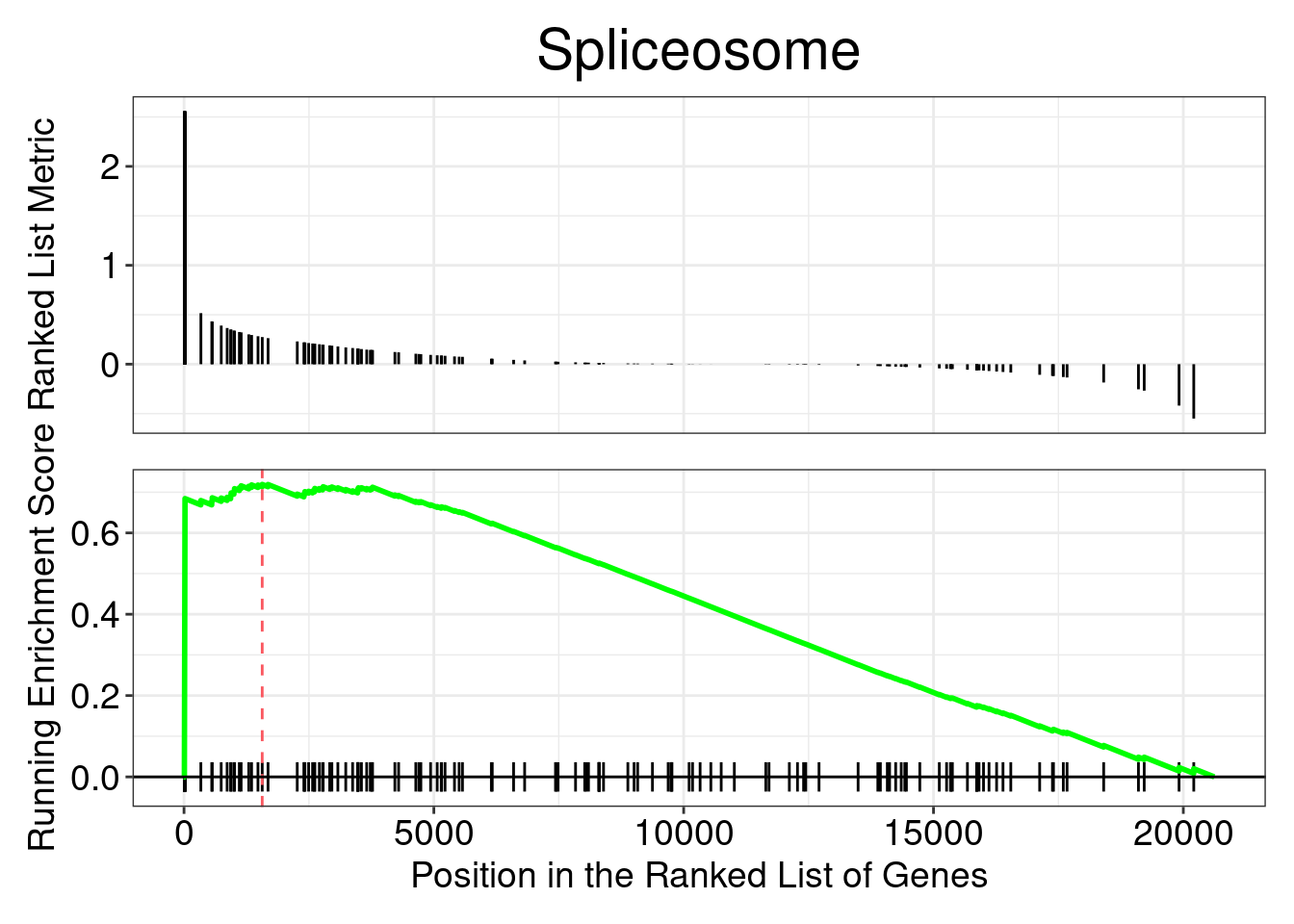
In this plot, the lines in plot represent the genes in the gene set, and where they occur among the log2 fold changes. The largest positive log2 fold changes are on the left-hand side of the plot, while the largest negative log2 fold changes are on the right. The top plot shows the magnitude of the log2 fold changes for each gene, while the bottom plot shows the running sum, with the enrichment score peaking at the red dotted line (which is among the negative log2 fold changes).
Use the Pathview R package to integrate the KEGG pathway data from clusterProfiler into pathway images:
## Output images for a single significant KEGG pathway
pathview(gene.data = foldchanges,
pathway.id = gseaKEGG_results$ID[1],
species = "hsa",
limit = list(gene = 2, # value gives the max/min limit for foldchanges
cpd = 1))NOTE: Printing out Pathview images for all significant pathways can be easily performed as follows:
## Output images for all significant KEGG pathways get_kegg_plots <- function(x) { pathview(gene.data = foldchanges, pathway.id = gseaKEGG_results$ID[x], species = "hsa", limit = list(gene = 2, cpd = 1)) } purrr::map(1:length(gseaKEGG_results$ID), get_kegg_plots)
Instead of exploring enrichment of KEGG gene sets, we can also explore the enrichment of BP Gene Ontology terms using gene set enrichment analysis:
# GSEA using gene sets associated with BP Gene Ontology terms
gseaGO <- gseGO(geneList = foldchanges,
OrgDb = org.Hs.eg.db,
ont = 'BP',
minGSSize = 20,
pvalueCutoff = 0.05,
verbose = FALSE)
gseaGO_results <- gseaGO@result
head(gseaGO_results)gseaplot(gseaGO, geneSetID = gseaGO_results$ID[1], title = gseaGO_results$Description[1])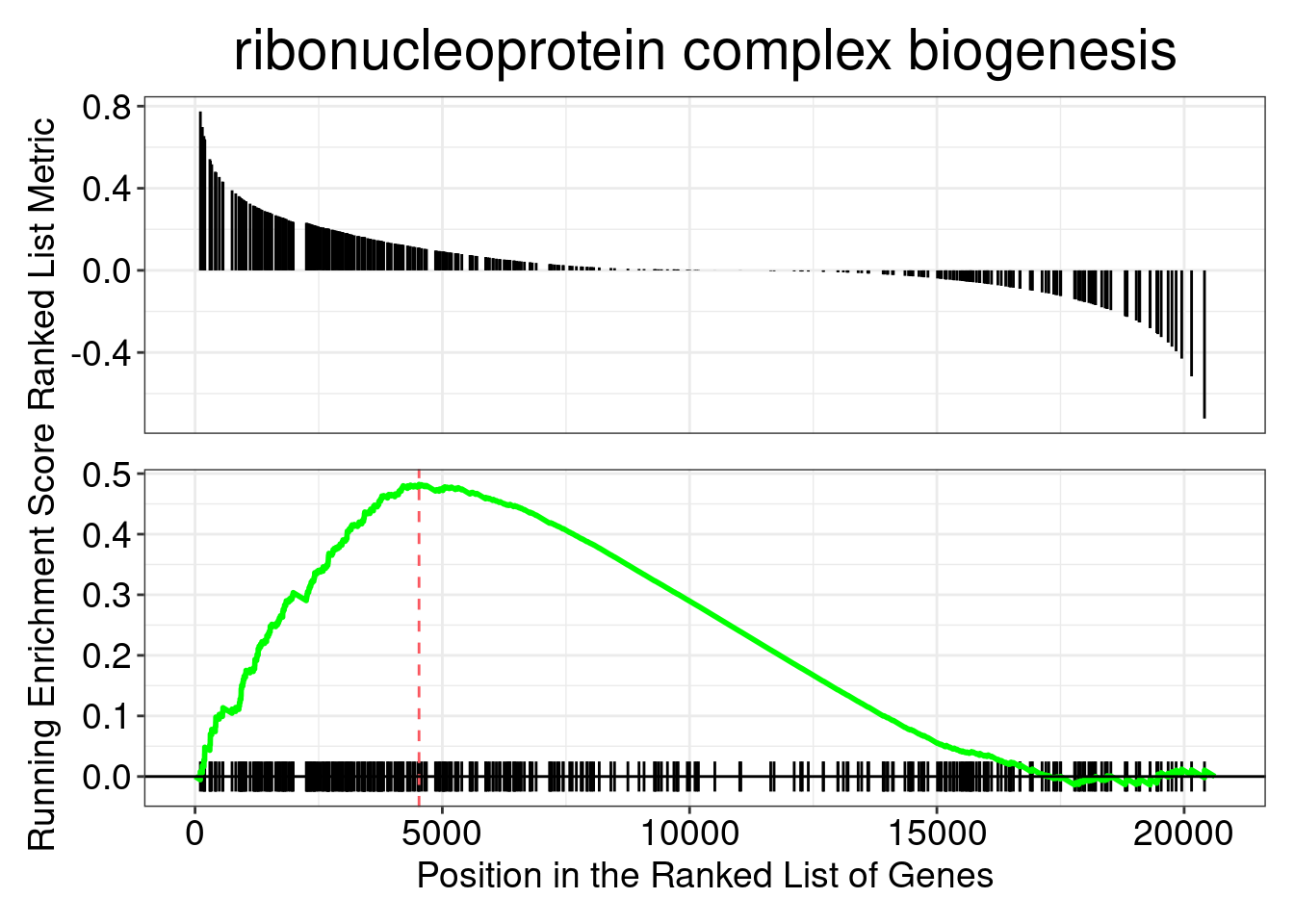
There are other gene sets available for GSEA analysis in clusterProfiler (Disease Ontology, Reactome pathways, etc.) You can check out this link for more!
Exercise 1
Run a Disease Ontology (DO) GSEA analysis using the gseDO() function. NOTE the arguments are very similar to the previous examples.
- Do you find anything interesting?
Exercise 2
Run an GSE on the results of the DEA for Garlicum vs Vampirium samples. Remember to use the annotated results!